- PHARMACOLOGIE CLINIQUE
- PHARMACOLOGIE CLINIQUELa pharmacologie clinique est la science qui étudie l’utilisation rationnelle des médicaments chez l’homme. Sous une forme plus moderne, elle a les mêmes objectifs que la thérapeutique médicamenteuse traditionnelle, mais elle en diffère essentiellement par les méthodes qu’elle utilise. Orientées vers des objectifs pharmacologiques précis, celles-ci sont à la fois très systématisées et de portée générale, s’appliquant pratiquement à toute nouvelle molécule susceptible de devenir un jour médicament. Elles permettent à chaque stade de l’étude puis du développement d’une nouvelle substance, depuis sa première utilisation chez l’homme, pendant sa commercialisation et même au-delà, de connaître les paramètres utiles à sa prescription.Les conditions de développement d’une activité optimale dans un contexte pathologique particulier et chez un individu déterminé peuvent être mesurées et souvent prévues par l’étude du métabolisme et de la pharmacocinétique du médicament. Les qualités respectives des différentes formes pharmaceutiques d’un même médicament sont appréciées par des études de biodisponibilité . L’étude clinique de l’activité pharmacodynamique et éventuellement des effets toxiques relève des essais thérapeutiques . Enfin, la pharmacovigilance permet de surveiller la stabilité ou l’évolution des effets connus des médicaments au cours de leur utilisation sur une grande échelle, sectorielle, nationale et internationale et, éventuellement, d’en découvrir d’autres.1. Métabolisme des médicamentsS’agissant d’un médicament donné, le terme de métabolisme désigne généralement la succession des phénomènes qui surviennent lorsque celui-ci est en contact avec l’organisme. Plus précisément, dans l’histoire biologique du médicament, le métabolisme implique ses différentes localisations, ses transformations éventuelles et les modalités de son élimination.Le métabolisme d’un médicament peut se décomposer en quatre phases: résorption, diffusion, transformations et élimination d’un médicament. Dans le devenir biologique elles n’ont cependant pas toutes la même importance. Pratiquement, deux étapes sont communes aux métabolismes de tous les médicaments: ce sont la diffusion et l’élimination. La résorption et les transformations n’existent que pour certaines voies d’administration et pour certaines molécules. On peut en effet éviter le temps de résorption en administrant le produit par voie intraveineuse: comme il s’agit en général d’un «temps mort», correspondant à la latence de l’effet recherché, celui-ci est alors supprimé, ce qui montre l’intérêt de cette voie d’administration en cas d’urgence.D’autre part, certains médicaments ne sont pratiquement pas transformés dans l’organisme; dans ces cas, l’élimination se fait exclusivement par voie rénale.La résorption , période initiale du métabolisme, correspond au passage du médicament dans le sang à partir de la forme pharmaceutique et de la voie d’administration utilisées. On décrit ainsi différents types de résorption: digestive, cutanée, musculaire, sous-cutanée, correspondant aux administrations orale, locale ou parentérale des formes pharmaceutiques correspondantes. La résorption est considérée comme totale ou partielle selon que la quantité administrée est ou non complètement résorbée.La diffusion est l’étape dans laquelle le médicament se répartit dans tout l’organisme par l’intermédiaire de la circulation sanguine. Qualitativement, il s’agit d’un phénomène général; tout tissu ou organe reçoit une fraction de la dose administrée, proportionnelle au débit de sang qui l’irrigue. Quantitativement, la répartition du médicament entre le sang et les différents tissus est fonction de leurs gradients de concentration, de leurs capacités respectives de fixation et de la liposolubilité de la molécule.Lorsque la capacité sanguine de fixation est la plus élevée, le médicament est, au moins en partie, retenu dans le sang, sa diffusion tissulaire est faible. En revanche, si elle est plus faible que les capacités tissulaires correspondantes, le médicament n’étant pas retenu dans le sang, sa diffusion tissulaire est grande. Sauf cas exceptionnels, les liaisons tant plasmatiques que tissulaires d’un médicament dans l’organisme sont des combinaisons réversibles, suivant la loi d’action de masse. Cela explique que les effets des médicaments comme leur présence dans l’organisme soient limités dans le temps (fig. 1).Le médicament diffuse dans tout l’organisme sous sa forme libre. Il est lié dans les tissus sous forme de combinaisons réversibles réalisant à chaque instant un état d’équilibre global, dont la durée dépend de la stabilité du médicament et de la rapidité de son élimination par les émonctoires.La transformation de la structure chimique du médicament par diverses réactions biochimiques conduit à la formation de substances appelées métabolites du médicament. Ceux-ci sont dits primaires s’ils résultent d’une seule réaction de transformation de la molécule initiale, secondaires lorsque deux réactions ont conduit à leur formation, tertiaires lorsque trois réactions de transformation sont nécessaires.Ces réactions se développent dans tout tissu pourvu de l’équipement enzymatique nécessaire; elles sont d’autant plus intenses que l’activité enzymatique y est plus grande: en pratique, c’est au niveau du foie, des reins, des poumons et de la peau que l’on observe la formation du plus grand nombre de métabolites. Certaines enzymes sont pharmacologiquement inductibles: l’administration de médicaments particuliers dits inducteurs augmente leur activité et, ainsi, le débit de métabolites formés.Selon l’activité pharmacologique de ces métabolites, les réactions de transformation peuvent provoquer soit une inactivation, soit au contraire une activation du médicament. Le plus souvent, les métabolites formés sont moins actifs que la molécule initiale, ce qui correspond à une limitation des effets médicamenteux. Dans tous les cas, cependant, les métabolites sont plus hydrosolubles que la molécule administrée, et ce fait facilite l’élimination du produit. La présence et l’importance de ces systèmes enzymatiques sont liées à des facteurs d’origine génétique. Il en résulte de très grandes variations individuelles, parfois qualitatives, toujours quantitatives dans les transformations métaboliques d’un médicament. Ce fait d’observation explique pourquoi l’efficacité d’un médicament et plus précisément l’intensité et la durée de ses effets peuvent être très différentes d’un individu à un autre en dehors de tout état pathologique.L’élimination est assurée principalement par le foie et les reins.Dans le premier cas, après fixation hépatique, ces substances sont sécrétées dans la bile, soit directement, soit après transformation. Lorsqu’il n’existe pas de résorption intestinale ultérieure (cycle entérohépatique), le médicament et/ou ses métabolites sont retrouvés dans les selles.Différents mécanismes expliquent l’élimination rénale des médicaments. Celle-ci peut être une simple filtration glomérulaire pour la substance sous forme libre, non liée aux protéines circulantes; elle peut aussi être accompagnée d’une sécrétion tubulaire. Le second mécanisme ne s’observe que pour les substances ionisées; il se réalise par transport actif, ce qui explique qu’il soit quantitativement limité tant par l’énergie que par les transporteurs qu’il nécessite. Le rôle du pH urinaire est évidemment fondamental puisqu’il conditionne le degré d’ionisation, donc de sécrétion des substances excrétées. En milieu acide, par exemple, les acides faibles, non ionisés, ne sont pas excrétés: leur présence et donc leurs effets sont prolongés. En milieu alcalin, en revanche, ils sont ionisés, leurs effets sont de plus courte durée. Un raisonnement analogue montre que, en milieu alcalin, les bases faibles agissent plus longtemps alors que c’est l’inverse après acidification des urines. On peut mettre à profit ces variations d’élimination urinaire soit en cas d’intoxication pour augmenter la vitesse d’excrétion d’une substance, soit, à l’inverse, pour augmenter la durée de ses effets.Il existe d’autres voies d’élimination d’un médicament et de ses métabolites. Ce sont essentiellement les voies pulmonaire (dans l’air expiré), cutanée (sueur), lacrymale (larmes). Elles sont en général quantitativement peu importantes. Une attention spéciale doit cependant être accordée à la sécrétion de médicament dans le lait maternel: elle peut, dans certains cas, réaliser un apport de médicament non négligeable au nouveau-né.2. PharmacocinétiqueLe déroulement des différentes phases du métabolisme n’est pas successif mais simultané; ainsi l’élimination commence sitôt qu’une quantité suffisante de médicament est résorbée. D’autre part, chacune d’elles peut s’exprimer sous forme d’équations mathématiques relativement simples, ce qui explique que l’on ait tenté de les quantifier. Cet aspect de la pharmacologie appartient à la pharmacocinétique .Cette discipline a pour objet le calcul des concentrations du médicament et de ses métabolites dans leurs différentes localisations et leur évolution au cours du temps qui suit l’administration du médicament. Ce calcul permet de prévoir, au moins en partie, les concentrations de la forme active d’un médicament obtenues par administration d’une dose donnée sous une forme pharmaceutique précise et par une voie déterminée, dans une ou dans plusieurs localisations. Il offre en outre la possibilité de déterminer un rythme d’administration de médicament tel qu’une imprégnation thérapeutique efficace soit maintenue sans surdosage ni inactivité. Enfin, lors d’états pathologiques graves où tout ou partie de ces fonctions métaboliques sont modifiées, il est un instrument précieux d’adaptation de la posologie.Les méthodes utilisées en pharmacocinétique reposent, chez l’homme, sur l’interprétation mathématique des courbes de décroissance plasmatique et d’apparition dans l’urine des médicaments après administration par différentes voies; en plus, chez l’animal, on utilise la mesure des concentrations tissulaires correspondantes.Considérons la courbe de décroissance des concentrations plasmatiques résultant de l’injection intraveineuse d’une dose de médicament: celle-ci peut s’exprimer par une ou plusieurs fonctions exponentielles selon l’équation générale suivante:
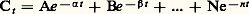 où Ct représente la concentration plasmatique à l’instant t , A et 見 les caractéristiques du premier compartiment, dit compartiment central, B et 廓 celles du compartiment suivant, N et n les caractéristiques du n -ième compartiment.L’analyse compartimentale attribue à chaque exponentielle la valeur d’un compartiment particulier, celui-ci étant défini de façon dynamique comme la masse de substance ayant, au même moment, un comportement identique dans l’organisme (fig. 2). Ainsi, selon la nature de la courbe de décroissance des concentrations plasmatiques et le nombre de processus exponentiels auxquels elle correspond, on distingue des cinétiques monocompartimentales, bicompartimentales ou multicompartimentales.Dans chaque compartiment, le devenir du médicament est caractérisé par son volume de distribution et sa demi-vie .Le premier est un volume apparent, théorique, sans correspondance anatomique ou physiologique. Il indique simplement le volume dans lequel la masse de médicament serait dissoute en l’absence de toute fixation: il permet d’avoir une idée de l’importance de la diffusion et de la fixation du médicament. La demi-vie correspondante indique le temps au bout duquel la concentration est devenue la moitié de la concentration initiale.Dans le cas d’un modèle à plusieurs compartiments, il est possible de calculer les constantes de transfert qui caractérisent l’intensité des flux de médicament entrant et sortant d’un compartiment donné. Un des cas les plus fréquemment rencontrés est celui d’un modèle ouvert à deux compartiments (fig. 2). Le compartiment central est celui où le médicament est injecté et d’où il est éliminé. Le compartiment périphérique (ou profond) représente les localisations tissulaires du médicament; son volume est plus grand, ses échanges avec le premier compartiment sont lents.Lorsque le médicament n’est pas administré par voie intraveineuse mais sous une forme qui doit être résorbée, on peut mesurer la constante de résorption correspondante. Celle-ci a l’intérêt de permettre l’appréciation de la durée de la résorption de la dose administrée. Lorsque celle-ci est lente, un effet retardé et prolongé est vraisemblable. En cas de résorption digestive, une vitesse trop lente peut cependant entraîner une résorption partielle (cf. infra , Biodisponibilité des formes pharmaceutiques ).Des différentes données pharmacocinétiques caractérisant un médicament, il est possible de tirer des renseignements importants concernant son utilisation chez l’homme. La notion d’un compartiment périphérique de grand volume est intéressante lorsqu’il s’agit par exemple de sélectionner un antibiotique destiné à traiter une localisation infectieuse profonde. Au contraire, la recherche d’un effet vasculaire orientera le choix du thérapeute vers des médicaments de faible volume de distribution.Avec la demi-vie d’élimination d’un médicament mesuré pour un individu déterminé, il est possible de calculer avec précision le rythme de son administration de manière à maintenir une imprégnation continue de l’organisme.On conçoit enfin que foie et reins étant les principaux organes d’élimination des médicaments, leur efficacité conditionne l’activité médicamenteuse. Toute atteinte fonctionnelle hépatique ou rénale peut ainsi modifier la cinétique et les caractéristiques des effets pharmacologiques attendus: il est possible de pallier cet inconvénient en connaissant la part respective de ces deux émonctoires dans le métabolisme des médicaments. Ainsi, dans l’insuffisance hépatique grave, on choisira de préférence des médicaments éliminés par les reins. En cas d’insuffisance rénale, on retiendra au contraire les substances dégradées par le foie.3. Biodisponibilité des formes pharmaceutiquesDiversité des formes pharmaceutiquesLe médicament, sous sa forme initiale purifiée, n’est pas directement utilisable en thérapeutique. En effet, il s’agit le plus souvent d’une poudre (digoxine, sulfate d’atropine) ou d’un liquide (clofibrate) dont l’administration en faible quantité est impossible ou très peu commode. On est donc obligé de l’administrer sous une forme pharmaceutique déterminée où l’on retrouvera l’exacte quantité que l’on souhaite délivrer au malade. Cette méthode suppose que la dose apportée soit aussi la dose active dans l’organisme. Or ce n’est pas toujours le cas, et différents travaux ont pu montrer qu’un même médicament, administré au même individu, à la même posologie, mais sous des formes pharmaceutiques différentes, pouvait développer des effets d’intensité variable. En d’autres termes, malgré des sujets et des posologies identiques, des formes pharmaceutiques différentes ne développaient pas le même effet. Cette observation a été faite en particulier avec des comprimés de digoxine de marques différentes, avec des gélules de chloramphénicol et de tétracycline. Elle s’explique par une inadéquation de la forme pharmaceutique, qui a deux causes principales. La première réside dans le fait que celle-ci peut ne libérer qu’une fraction du médicament qu’elle contient: la quantité résorbée est donc inférieure à la dose administrée (cf. chap. 1 et 2), la fraction restante étant éliminée dans les selles. Dans d’autres cas, la forme pharmaceutique libère bien la totalité du médicament qu’elle contient, mais avec une vitesse si lente que les concentrations plasmatiques correspondantes (après résorption) seront toujours faibles. Le gradient de concentration entre le plasma et les tissus le sera également: la diffusion tissulaire sera peu importante, l’effet pharmacologique résultant sera faible, voire nul.On comprend ainsi qu’après avoir sélectionné un médicament il est absolument indispensable de l’administrer sous une forme pharmaceutique telle qu’il puisse, à partir d’elle, développer la totalité des effets que l’on attend de lui. C’est de cette constatation qu’est née la biopharmacie , discipline qui étudie les relations qui existent entre les propriétés physico-chimiques du médicament et de ses formes pharmaceutiques et les effets pharmacologiques qu’ils produisent. La notion de biodisponibilité ou disponibilité biologique vise à corréler la dose administrée à l’effet qu’elle produit lorsqu’elle est utilisée sous une forme pharmaceutique résorbable. La notion de bioéquivalence envisage l’efficacité relative d’un même médicament, administré à la même posologie, sous des formes pharmaceutiques différentes. Les formes pharmaceutiques sont bioéquivalentes si elles produisent des effets d’intensité et de durée identiques; dans le cas contraire, elles ne le sont pas et alors il est important de ne pas remplacer l’une par l’autre.Paramètres de biodisponibilitéThéoriquement, la biodisponibilité d’une forme pharmaceutique devrait être définie par deux paramètres: la quantité de médicament mise à la disposition des sites d’action et la vitesse de cette mise à disposition. Or, cette définition théorique est peu réaliste. En effet, quantifier ces deux paramètres au niveau du site d’action est difficilement réalisable chez l’homme. En revanche, la détermination de ces paramètres peut se faire plus aisément si l’on considère non plus les sites d’action, mais la quantité de médicament présente dans la circulation sanguine. On mesure alors une biodisponibilité systémique . De nombreux travaux ont en effet montré que, pour un grand nombre de médicaments, il existe une relation entre leurs concentrations sanguines et leurs effets pharmacologiques tissulaires; la concentration sanguine, en d’autre termes, est dans une certaine mesure le reflet de la concentration au niveau du site d’action induisant l’effet pharmacologique. D’où la définition suivante pratiquement admise par tous: la biodisponibilité d’une forme pharmaceutique représente la fraction de médicament administré qui atteint la circulation générale et la vitesse avec laquelle elle l’atteint. Par définition, le médicament contenu dans une forme pharmaceutique administrée par voie intraveineuse a une biodisponibilité de 100 p. 100, puisque, dans ces conditions, on évite l’étape de la résorption qui est donc à la fois totale et instantanée. Administré par une autre voie (orale, rectale, sublinguale, etc.), le médicament va devoir traverser un certain nombre de barrières biologiques qui peuvent représenter une limitation de son passage dans la circulation sanguine. De ce fait, la biodisponibilité s’exprimera alors par comparaisons. On distingue ainsi:– la biodisponibilité absolue. Elle est évaluée par comparaison avec la voie intraveineuse. Par exemple, on évalue la biodisponibilité absolue d’un comprimé de digoxine en comparant les concentrations sanguines de digoxine obtenues au cours du temps après administration orale avec celles obtenues après injection intraveineuse de la même dose;– la biodisponibilité relative. Dans ce cas, on administre une forme pharmaceutique de biodisponibilité connue, dite forme de référence, par une voie autre que la voie intraveineuse. Lorsqu’on compare la biodisponibilité de différents comprimés contenant le même médicament, dont la mise en forme pharmaceutique a été différente, celui d’entre eux présentant la meilleure biodisponibilité relative exprimera la biodisponibilité relative optimale.Facteurs de biodisponibilitéLes facteurs influençant la biodisponibilité d’une forme pharmaceutique sont multiples. Les caractères physico-chimiques de la molécule peuvent être d’abord incriminés: ils conditionnent sa solubilité et donc sa vitesse de dissolution ainsi que sa résorption. Or, pour être résorbé, un médicament doit d’abord être solubilisé (fig. 3). Sa vitesse de dissolution dépend de ses formes cristallines et/ou chimiques (estérification, salification, état d’hydratation); elle est généralement fonction de la taille des particules et de l’état d’ionisation de la molécule. De même, pour la fabrication de la forme pharmaceutique, le médicament doit être mélangé à divers excipients, c’est-à-dire à des substances inactives par elles-mêmes qui facilitent son administration et sa conservation. Certains excipients ont tendance à absorber une fraction du médicament de sorte que la quantité parvenant dans la circulation générale est alors moindre. D’autres modifient la vitesse de dissolution du médicament, réduisant ou augmentant ainsi sa biodisponibilité. Les conditions de fabrication peuvent également influencer la biodisponibilité: lors de la fabrication d’un comprimé, la compression du mélange à mettre en forme, pouvant légèrement varier d’un jour à l’autre, modifiera la vitesse de dissolution. Enfin, les conditions de conservation de la forme pharmaceutique peuvent être responsables de la dégradation chimique ou d’une modification cristalline et entraîner ainsi une évolution de la biodisponibilité.Mesure de la biodisponibilitéLa biodisponibilité d’une forme pharmaceutique peut être déterminée soit indirectement, par la mesure de l’effet qu’elle produit, soit directement, en mesurant la quantité contenue dans l’organisme.La première méthode est intéressante dans la mesure où elle permet de mesurer directement la bioéquivalence des deux formes pharmaceutiques d’un même médicament. Elle se heurte malheureusement à la difficulté de mesurer avec précision l’intensité de certains effets thérapeutiques et, de ce fait, n’est que rarement employée.Le deuxième groupe de méthodes fait appel à l’analyse pharmacocinétique du médicament dans les différents liquides biologiques (sang et/ou urines) après administration unique ou répétée. La détermination de la quantité de médicament résorbée par la mesure des taux sanguins repose sur la loi de Dost (1964) ou principe des aires correspondantes: «Quelle que soit la voie d’administration et quel que soit le nombre de compartiments de distribution dans l’organisme, la quantité résorbée est toujours proportionnelle à l’aire sous la courbe des concentrations sanguines extrapolée de 0 à l’infini.» Pour être utilisable, ce principe impose trois contraintes principales:– il doit exister dans l’organisme un compartiment central incluant le sang à partir duquel se produit l’élimination ; l’expérience a montré que ce postulat est vérifié dans la très grande majorité des cas;– la cinétique de l’élimination du médicament doit être de premier ordre; cette condition est souvent réalisée, mais ce n’est pas toujours le cas, et il faut toujours la vérifier;– le volume du compartiment central et la constante de vitesse globale d’élimination (ke ) restent constants au cours des essais; ce dernier point n’est jamais rempli.Il est donc nécessaire de mesurer ces paramètres et de corriger l’aire sous la courbe en fonction de leurs valeurs respectives au moment de l’étude. À partir de ce principe, si ces conditions sont remplies, il est donc possible de mesurer la biodisponibilité absolue ou relative d’une forme pharmaceutique par comparaison des surfaces sous la courbe correspondant aux différentes formes. Cette surface peut être calculée après administration unique ou lors d’une administration chronique, entre deux administrations. Parallèlement à la quantité globale de médicament résorbé, on peut également mesurer la vitesse de résorption à partir des taux plasmatiques. Il est en effet possible, à partir d’une étude pharmacocinétique des taux plasmatiques observés, de calculer la constante de résorption selon un modèle compartimental. Mais il est aussi possible de mesurer simplement la concentration plasmatique maximale et le temps nécessaire pour l’obtenir après l’administration. Ces deux paramètres sont en effet directement liés à la vitesse de résorption; lorsqu’elle diminue, la concentration plasmatique maximale diminue et le temps nécessaire pour y parvenir augmente, et réciproquement. Les mêmes renseignements peuvent être retrouvés à partir des concentrations urinaires. En effet, si un médicament est éliminé uniquement dans les urines et sans transformation, la quantité éliminée est alors égale à la quantité résorbée. On peut ainsi dans ce cas particulier évaluer la biodisponibilité absolue sans recourir à l’administration intraveineuse. Si le médicament est métabolisé, il en sera de même si l’on connaît tous les métabolites et que l’on est capable de mesurer leur excrétion. L’analyse pharmacocinétique des données urinaires peut également fournir des renseignements concernant la vitesse de résorption du médicament, mais cette analyse est généralement plus difficile à réaliser et moins précise que l’étude des concentrations plasmatiques.Enfin, de nombreuses études ont permis de corréler la biodisponibilité d’une forme pharmaceutique in vivo chez l’homme avec des tests de dissolution in vitro . Il n’a pas été possible à ce jour de dégager une relation générale, mais ce type d’essai in vitro peut être utilisé pour vérifier l’égalité de biodisponibilité entre deux lots de fabrication, à la condition que le test in vitro ait été étalonné au préalable vis-à-vis de la biodisponibilité in vivo .Problèmes pratiquesIl est nécessaire de définir les critères de jugement concernant la biodisponibilité d’une ou de plusieurs formes pharmaceutiques. Deux différentes approches peuvent être utilisées. D’un point de vue mathématique, l’analyse statistique à l’aide de tests paramétriques ou non paramétriques permet avec un risque d’erreur connu de décider si deux formes pharmaceutiques sont différentes ou non et de calculer un intervalle de confiance correspondant. Mais, d’un point de vue clinique, il est également nécessaire de considérer la variation d’effets qui peuvent être engendrés. Ainsi, on s’accorde généralement pour considérer que deux médicaments sont bioéquivalents si leur biodisponibilité ne diffère pas de plus de 20 p. 100. Mais ce critère ne peut être le seul à considérer. Si, en effet, on se trouve en présence de deux formes, l’une ayant une biodisponibilité de 50 p. 100 avec une variabilité de 10 p. 100, l’autre de 75 p. 100 avec une variabilité de 25 p. 100, il sera préférable d’utiliser la première forme, bien que sa biodisponibilité soit plus faible. En effet, la réponse thérapeutique sera alors beaucoup plus reproductible et cette forme pharmaceutique plus maniable.Les études de biodisponibilité, requises pour commercialiser une nouvelle forme pharmaceutique (étude in vivo ) et pour s’assurer de l’adéquation de tout nouveau lot de fabrication (étude in vitro ), permettent de vérifier et de garantir la bioéquivalence des préparations d’un même médicament mises à la disposition du praticien. Si elles apportent une grande sécurité sur le plan de la fiabilité de ces formes pharmaceutiques, elles ne peuvent cependant garantir de façon absolue l’efficacité des médicaments qu’elles contiennent. Tous les autres facteurs propres au sujet traité demeurent: ainsi, tout trouble pathologique de la résorption intestinale peut diminuer la quantité de médicament résorbée, même si la forme pharmaceutique utilisée a une excellente biodisponibilité. Plus simplement, l’apport simultané de plusieurs médicaments, la prise d’aliments peuvent modifier quantité résorbée et vitesse de résorption de l’un ou de plusieurs d’entre eux. L’étude de biodisponibilité n’est plus en cause dans ces cas: elle apporte seulement, et c’est déjà un intérêt considérable, la preuve que les formes pharmaceutiques sont adéquates. La modification pharmacologique observée relève alors d’une autre cause, d’ordre individuel, qu’il convient de déterminer.4. Essais thérapeutiquesQuelle que soit la qualité des travaux effectués lors de l’étude préclinique d’un nouveau médicament, les résultats obtenus sur des animaux n’ont qu’une valeur d’orientation prévisionnelle des effets que l’on pourra observer chez l’homme. La première fois qu’un nouveau médicament est administré à un homme, il s’agit donc bien d’une expérimentation. Cela explique les réticences d’ordre éthique qui sont ressenties par tous médecins, légistes, moralistes, patients...Cependant, les progrès réalisés au cours des dernières décennies dans le traitement de nombreuses maladies sont une raison suffisante et une justification à persévérer dans la voie des essais puisque ces traitements nécessitent soit une amélioration des médicaments utilisés pour les traiter, soit la découverte de nouveaux produits efficaces. Les recherches se poursuivant activement aux niveaux chimique et pharmacologique préclinique, il y aura donc toujours dans l’histoire naturelle de ces nouveaux médicaments, actuels ou futurs, un premier passage sur l’homme.Les règles de l’éthique médicale imposent de ne faire ces essais que dans des conditions strictement définies: les risques, inhérents à toute nouvelle thérapeutique, doivent être largement compensés par les bénéfices escomptés. Les essais doivent être parfaitement codifiés afin que les résultats obtenus puissent être interprétés sans équivoque. Toutes les garanties de surveillance par des équipes médicales, compétentes et entraînées, doivent être données aux sujets qui recevront le nouveau médicament.Phases de l’essai clinique du médicamentLa nécessité d’une expérimentation parfaitement planifiée, garantissant le minimum de risques aux sujets recevant le nouveau médicament, s’est fait jour dans l’esprit des cliniciens il y a plusieurs décennies, mais ce n’est que depuis peu que l’investigation clinique d’un nouveau médicament a été divisée en plusieurs phases. Codifiées à partir de données des études de pharmacologie préclinique ou animale, les phases, au nombre de quatre dans la classification de la F.D.A. (Food and Drug Administration), sont telles que la progression d’une phase à l’autre est conditionnée par les résultats obtenus à chaque étape.Phase I, ou première administration à l’hommeAu cours de cette phase, sont étudiés l’activité biologique générale, la pharmacocinétique, le métabolisme et la tolérance du nouveau produit. Ces divers éléments sont étudiés sur un petit nombre de sujets volontaires sains. Le problème juridique du volontaire sain recevant un nouveau médicament est résolu dans la majorité des pays, notamment ceux de l’Union européenne. À la suite de la phase I qui dure de un an à un an et demi, la tolérance de l’organisme au produit nouveau étant définie, la connaissance de ses caractéristiques pharmacocinétiques permet de proposer une posologie , c’est-à-dire une quantité de médicament qui, administrée selon un certain rythme, aura un maximum de chances d’entraîner un effet thérapeutique, sans effet toxique. C’est également au cours de cette phase I que certaines interactions peuvent être étudiées: par exemple l’interaction de l’alcool avec les anxiolytiques.Phase IIAu cours de la phase I, l’activité thérapeutique , c’est-à-dire la possibilité de traiter une maladie ou un symptôme, n’a pas du tout été abordée. Pendant la phase II, qu’on subdivise parfois assez arbitrairement en phase II précoce et en phase II tardive, auront lieu des essais cliniques , sur des malades sélectionnés, pendant une période allant de un à deux ans. Les études de la phase II s’appuient sur les résultats obtenus en phase I. Elles vont permettre de définir:– la validité des posologies proposées;– l’existence d’une relation entre le médicament et les effets thérapeutiques: relation dose-effet ou relation concentration plasmatique-effet.S’adressant à des sujets malades, on étudie également au cours de cette phase l’influence de la maladie sur le métabolisme et la pharmacocinétique du nouveau médicament. On peut aussi observer dès cette phase l’existence d’interactions médicamenteuses. Les recherches effectuées en phase II le sont dans des services hospitaliers hautement spécialisés. Les réactions des malades sont suivies de très près par des médecins spécialistes, ce qui assure aux malades les garanties le plus complètes possible de sécurité.Phase IIILes résultats des phases I et II permettent de décider de la poursuite ou de l’arrêt de l’étude du nouveau produit. En cas de poursuite, la phase III permet, en se plaçant dans les conditions le plus proches possible de la pratique médicale quotidienne, de confirmer les informations obtenues au cours des phases précédentes et de définir les conditions exactes de l’utilisation du nouveau produit. On définit ainsi les indications et les contre-indications d’utilisation dans l’arsenal thérapeutique disponible. Cette phase est, par ses techniques et méthodologies, proche de la phase II. Elle en diffère cependant par l’utilisation de grandes séries de malades traités (essais contrôlés), ce qui nécessite une très grande rigueur expérimentale, afin de permettre un traitement statistique correct des informations. Elle ne nécessite pas, par contre, de services médicaux hautement spécialisés puisque, en phase II, les inconvénients majeurs qui pourraient être liés au nouveau médicament ont été détectés. Elle aboutit à l’autorisation de mise sur le marché (A.M.M.).Phase IVElle suit la mise du nouveau médicament sur le marché. Elle n’appartient plus tout à fait à une phase d’«essais» cliniques, mais elle est pourtant indispensable pour mettre en évidence les effets médicamenteux indésirables qui, compte tenu de la durée relativement courte des phases I, II et III, n’auraient pas pu apparaître.C’est également durant cette phase que seront complétées les informations concernant des médicaments qui sont administrés au long cours, pour des maladies chroniques de longue durée. Il faut également rappeler que c’est à partir d’observations effectuées par des cliniciens curieux, en phase IV, que de nouvelles indications thérapeutiques peuvent apparaître. À ce stade, on recherche, après les effets indésirables, les indications nouvelles, interactions particulières. Enfin, cette phase est celle au cours de laquelle les rapports risque-bénéfice et coût-bénéfice pourront être établis.Techniques d’études utilisées lors des essaisIl existe deux grands types de méthodes: les essais dits ouverts, les essais dits contrôlés. Nous allons rapidement les passer en revue en essayant de dégager les avantages et les inconvénients de chacune de ces techniques et en précisant leur place dans les différentes phases. Quelle que soit la méthode utilisée, elle nécessite une équipe médicale parfaitement entraînée et attentive, tout au long de l’essai, à suivre les étapes du plan expérimental établi.Ce plan expérimental devra, lui, être soumis à des règles strictes d’éthique qui garantiront dans tous les cas une parfaite sécurité au malade. La tendance actuelle est de soumettre les plans expérimentaux d’essais thérapeutiques à des comités d’éthique locaux, régionaux ou nationaux. Le Code d’éthique s’appuie sur les déclarations successives faites à Nuremberg (1947), à Helsinki (1964) et à T 拏ky 拏 (1977), sur les règles à suivre concernant les expérimentations cliniques, c’est-à-dire faites sur des sujets humains. La phrase introductrice de la déclaration de T 拏ky 拏 fixait la limitation de la poursuite de l’essai thérapeutique chez un sujet donné: «La mission du médecin est de veiller à la santé de l’homme.» Elle poursuivait: le serment de Genève engage le médecin à «considérer la santé du patient comme son premier souci», et le Code international d’éthique médicale «interdit au médecin de donner un conseil ou de poser un acte médical prophylactique, diagnostique ou thérapeutique qui ne soit pas justifié par l’intérêt direct du patient et notamment d’affaiblir la résistance physique ou mentale d’un être humain, à moins de nécessité thérapeutique». Dans cet esprit, la loi Huriet sur la recherche biomédicale fut adoptée par le Parlement français en décembre 1988.Essai ouvertLe principe en est simple. Le malade et le médecin connaissent le traitement utilisé. L’expérimentateur mesure les effets obtenus, soit par l’observation clinique, soit par des mesures biologiques. Il importe d’utiliser un grand nombre de sujets, car l’élément subjectif de la thérapeutique demeure entier tant du côté du malade que du côté du médecin. Le traitement statistique essaiera de mettre en évidence l’efficacité du traitement. Cette technique est le plus souvent utilisée lors des phases IV des essais. Ce type d’essais présente deux avantages dont le premier est la simplicité, la souplesse. Le second avantage réside dans la possibilité de modifier la thérapeutique à tout instant selon les réactions du malade. Cette méthode est parfaitement applicable quand on possède des moyens objectifs de mesurer l’effet du médicament. Elle est pratiquement la seule applicable pour des essais de très longue durée.Il existe également des inconvénients qui limitent l’extension de ce type de technique. Le premier est l’impossibilité de mesurer objectivement l’effet du médicament dans maintes maladies. En second lieu, il importe de ne pas méconnaître l’aspect psychologique de la prescription: connaissant le médicament prescrit, le médecin peut influencer la réaction du malade par son seul comportement. Le malade lui-même peut réagir de façon orientée, simplement parce qu’il sait recevoir un médicament nouveau.Essais contrôlésLes difficultés posées par la mesure objective des effets des médicaments, l’importance de la composante psychologique, la nécessité de comparer les effets obtenus avec un nouveau médicament, soit à un placebo, soit à un médicament connu, ont fait se développer les techniques dites d’essais contrôlés. Nous décrirons les trois méthodes principales des essais contrôlés.Méthode du simple aveugleLe médecin connaît, le malade ignore quel type de produit est administré. On supprime dans ce cas la composante psychologique du malade. Selon la maladie traitée et les possibilités techniques, on utilise simultanément le médicament nouveau et un médicament de référence ou un placebo. Pour tous les cas, les voies et le rythme d’administration sont parfaitement superposables. Les malades sont répartis au hasard en deux groupes, un groupe recevant le placebo ou le produit de référence, l’autre groupe recevant le produit nouveau. Dans cette méthode, si la composante psychologique du malade est diminuée, celle du médecin persiste. Applicable en phases II et III, seule à pouvoir être utilisée dans certains cas, c’est lorsqu’elle est associée à une possibilité de mesure objective de l’effet du médicament qu’elle prend toute sa valeur, car elle laisse au médecin toute liberté de modifier la prescription, comme il en serait si le médicament n’était pas en expérimentation. Elle permet en d’autres termes d’être proche d’une situation thérapeutique normale.Méthode du double aveugleLorsque l’impossibilité d’une mesure objective de l’effet thérapeutique ou le biais possible, lié au rôle psychologique du médecin, sont susceptibles de fausser les résultats, on emploie la méthode dite du double aveugle. Dans ce cas, le malade et le médecin prescripteur ignorent la qualité du produit administré. Comme dans la méthode précédente, la population tirée au sort a été séparée en deux groupes, l’un recevant le médicament témoin ou le placebo, l’autre, le produit nouveau. L’avantage majeur de cette technique réside dans la suppression complète des facteurs subjectifs d’appréciation de l’effet thérapeutique.L’inconvénient majeur est l’aspect artificiel de la situation thérapeutique. En effet, la sélection, la distribution au hasard de la thérapeutique, la non-connaissance par le médecin prescripteur de ce qu’il a prescrit est une situation qui n’existe pas en dehors de l’essai. Il devient difficile pour le médecin prescripteur d’adapter la thérapeutique à l’évolution de la maladie. Malgré tous ces inconvénients, la méthode du double aveugle est actuellement la méthode qui semble donner les meilleurs résultats pour l’essai de nouveaux médicaments, surtout quand il n’existe pas de méthodes objectives permettant de mesurer, de quantifier, l’effet thérapeutique.Méthode de l’analyse séquentielleCette méthode est une variante de la méthode du double aveugle. Elle permet, sur un plus petit nombre de malades et dans un délai de temps court, d’obtenir une réponse suffisamment précise, au début d’une phase III, pour confirmer l’intérêt que peut présenter le nouveau médicament. Les malades se servant de témoins à eux-mêmes reçoivent successivement le produit à l’essai et le produit de référence ou le placebo. On peut également apparier les malades, l’un servant de témoin à l’autre. On a préalablement pris le soin de définir statistiquement, sur un graphique, l’évolution probable en présence d’un traitement efficace, et on a défini les frontières de l’efficacité et de l’inefficacité. On porte ensuite sur ce graphique l’évolution des grandeurs biologiques ou cliniques mesurées, qui traduisent l’effet du traitement. Quand, au cours de l’évolution, la ligne coupe l’une ou l’autre des lignes préalablement tracées, on a la réponse à la question posée. L’inconvénient majeur de cette technique est qu’elle n’est applicable que dans les cas où l’on possède un élément simple et rapidement mesurable pour juger de l’effet de la thérapeutique.Autres méthodesNous avons à plusieurs reprises évoqué l’utilité d’une mesure objective de l’effet du médicament. Chaque fois que cette possibilité existe, associée et corrélée à la mesure des concentrations plasmatiques du nouveau médicament, elle remplit toutes les conditions d’un essai contrôlé. De plus, elle permet une surveillance thérapeutique réelle, identique à celle qui peut être exercée en dehors de l’essai clinique du nouveau médicament.Bilan de l’expérimentationIl ressort, à l’évidence, de ce qui précède que l’essai thérapeutique nécessite des techniques expérimentales dont la rigueur doit être au moins égale, sinon supérieure, aux techniques utilisées sur l’animal. Il est faux de croire que les impératifs éthiques de l’essai du médicament limitent la rigueur de l’expérimentation. Cette rigueur est l’une des composantes essentielles des précautions à prendre pour l’essai du médicament chez l’homme. Elle seule permet de traiter un malade, alors même qu’il reçoit un nouveau médicament. Le développement des méthodes modernes d’exploration diagnostique chez l’homme permet de développer cette rigueur, cette objectivité dans l’appréciation des effets.Le développement des méthodes statistiques spécifiquement appliquées à l’essai thérapeutique a permis de traiter correctement les résultats obtenus. Il reste le problème de l’appréciation du rapport risque/bénéfice. Il est des cas, favorables, où, à la suite des phases II et III de l’expérimentation, on a pu évaluer le rapport risque/bénéfice. Il est d’autres cas moins favorables où seule la phase IV permettra, à partir d’un grand nombre de malades traités pendant une longue période, d’évaluer ce rapport. Enfin, il ne faut pas oublier que, dans de nombreux cas, la connaissance physio-pathologique fragmentaire de la maladie traitée ne permet pas d’apprécier objectivement l’effet d’une nouvelle thérapeutique. Il reste dans ce domaine de nombreux progrès à réaliser qui permettront une amélioration des essais thérapeutiques.L’essai thérapeutique contribue par lui-même à améliorer la connaissance fondamentale du mécanisme des maladies et de l’effet des thérapeutiques, cela grâce à la rigueur introduite dans l’expérimentation. Cette étude doit être considérée comme une recherche fondamentale et doit bénéficier des mêmes efforts que d’autres recherches fondamentales.Enfin, c’est probablement lors de ces essais thérapeutiques que la relation entre le médecin et le malade peut s’exprimer avec le plus de force. La confiance du malade pour son médecin doit, en effet, être totale, et l’attention du médecin pour son malade doit être, s’il se peut, encore plus complète au cours de cet acte thérapeutique privilégié.5. PharmacovigilanceDéfinitionsLe médicament, c’est «toute substance ou produit utilisé ou destiné à être utilisé en vue de modifier ou d’étudier un système physiologique ou un état pathologique, dans l’intérêt du sujet auquel il est administré».La réaction adverse, c’est toute «réaction nocive, non recherchée, qui apparaît aux doses normalement utilisées chez l’homme», lors de l’utilisation thérapeutique d’un médicament.La pharmacovigilance (Adverse Drug Monitoring) a pour tâche d’«obtenir des indications systématiques sur les liens de causalité probables entre médicaments et réactions adverses dans une population». Son but est d’établir la liste des effets adverses des médicaments, c’est-à-dire de connaître de la façon le plus précise possible les effets dits secondaires, latéraux, imprévus ou indésirables (terme actuellement préféré en France), tous effets qui se développent parallèlement à l’effet thérapeutique recherché. Le travail de pharmacovigilance se situe après l’apparition de l’effet indésirable. Celui-ci est d’abord notifié par le clinicien qui l’a observé, il est ensuite étudié par l’équipe de pharmacovigilance, qui l’enregistre, en fait une première évaluation et le transmet aux instances nationales. Ce travail, réalisé à l’échelon national puis international, aboutit à une somme d’informations permettant de connaître tous les effets adverses d’un médicament et leurs fréquences relatives. Cette méthode s’apparente beaucoup aux techniques épidémiologiques, ce qui explique que l’on ait pu définir la pharmacovigilance comme l’«épidémiologie des effets adverses des médicaments». On comprend alors l’importance fondamentale de l’aspect systématique du recueil des données. C’est seulement si tous les cas d’effets adverses sont enregistrés que leur incidence sera connue avec précision. Les définitions du médicament et de la réaction adverse permettent de préciser le champ d’action de la pharmacovigilance.Les médicaments suivis sont déjà commercialisés; délivrés avec ou sans ordonnance; utilisés à des doses convenables; prescrits dans une indication convenable.C’est ainsi que les médicaments en cours d’expérimentation (expertises cliniques) ne relèvent pas actuellement de la pharmacovigilance. De même, les effets de doses toxiques ou résultant d’indications non thérapeutiques en sont écartés: ils relèvent de la toxicologie.Par rapport à ces textes, nous avons pris l’initiative de recueillir, outre les effets adverses, les effets imprévus mais bénéfiques (type action du clofibrate dans le diabète insipide). Bien que ceux-ci soient rares, ils sont en effet importants à connaître rapidement et peuvent représenter un atout supplémentaire pour le médicament dans une prescription ultérieure.Quelques particularités d’effets indésirablesIl peut paraître choquant d’admettre que les substances utilisées en thérapeutique, considérées comme des médicaments, reconnues officiellement comme tels, aient encore besoin d’être étudiées. Ne peut-on prévoir de façon complète et définitive les effets des médicaments avant leur commercialisation? Différents arguments permettent d’affirmer qu’un tel objectif relève de l’utopie. En effet, si certaines réactions adverses sont relativement simples à déceler, d’autres sont beaucoup plus difficiles à mettre en évidence.Ce sont d’abord les réactions adverses rares. Leurs fréquences sont de l’ordre de 10-6 ou 10-9. Or une étude préalable, même extrêmement approfondie, ne peut porter sur 106 ou 109 sujets; en outre, l’apparition d’un seul cas d’effet nocif dans la totalité de la population étudiée est difficilement interprétable en tant qu’effet adverse. Un tel effet est donc pratiquement imprévisible. Un exemple nous permettra de préciser cet aspect de certaines réactions adverses. La digitaline est utilisée en thérapeutique depuis 1785, d’abord sous forme de préparations de digitale, puis sous forme cristallisée (1868): c’est donc un très vieux médicament qui, entre autres effets adverses, peut provoquer des thrombocytopénies. À l’heure actuelle, on en connaît environ neuf cas, ce qui représente une fréquence de survenue extrêmement faible, compte tenu de l’utilisation de ce médicament. En supposant que la digitaline soit aujourd’hui en cours d’expérimentation, devrait-on attendre et prolonger celle-ci jusqu’à la caractérisation indiscutable des thrombocytopénies? Évidemment non, et devant l’intérêt que présentait cette molécule lors de ses premières utilisations, le risque plaquettaire, ignoré à l’époque, nous semble bien minime. Nous retrouvons ici la notion générale de risque thérapeutique inhérente à l’utilisation de tout médicament, qu’il faut prendre sans hésitation lorsque le bénéfice thérapeutique est évident, mais qui impose une surveillance constante (fig. 4).Les réactions adverses peuvent, dans certains cas, avoir une latence de survenue extrêmement longue, bien supérieure au temps des essais. Dans ces conditions, il est bien évident que seule la surveillance constante du médicament au cours de sa commercialisation permet de les dépister. Les exemples de ce type sont surtout ceux d’effets toxiques. Le cas des sels de thorium (radioactifs) utilisés comme opacifiants biliaires en 1930 est classique; ils peuvent entraîner, environ dix à quinze ans après une seule administration, l’apparition d’une atteinte néoplasique du foie et des voies biliaires ou d’une leucémie. Il est bien évident que si l’utilisation en clinique humaine de telles substances était plausible en 1930, elle ne le serait plus du tout aujourd’hui.En effet, d’une part, on n’utilise plus de substances radioactives de ce type, car leurs périodes sont trop longues (1,4 憐 1010 années pour le thorium), d’autre part, une étude métabolique (obligatoire actuellement) aurait montré que ces substances se fixent de façon pratiquement irréversible dans l’organisme, ce qui exclut tout intérêt thérapeutique. Cet exemple prouve cependant qu’il est possible d’observer aujourd’hui les effets d’une substance qui n’est pourtant plus utilisée depuis longtemps.Les réactions adverses peuvent aussi apparaître soudainement au cours de l’utilisation thérapeutique du médicament. Ces cas se différencient des précédents car ils ne peuvent être attribués à des latences de longue durée. L’exemple le plus actuel est celui des sels de bismuth qui, utilisés depuis le début du XIXe siècle, provoquent depuis environ cinq ans un nombre non négligeable d’encéphalopathies. Ces nouveaux effets adverses ont été signalés en Australie en 1973, puis en France en 1974. Un tel phénomème montre clairement la nécessité d’une surveillance constante de la population qui reçoit des médicaments.Comment expliquer l’extrême rareté de certains effets adverses comme les thrombocytopénies des digitaliques? Quelles sont les causes de l’apparition brutale et récente des encéphalopathies aux sels de bismuth?Dans le premier cas, il s’agit vraisemblablement de réactions immunologiques; dans le second, une «adaptation» récente de certaines bactéries intestinales aux sels de bismuth permettant la synthèse in situ de métabolites résorbables et toxiques a été invoquée. Dans les deux cas, pratiquement, on constate que ce sont moins les médicaments que leurs récepteurs – sujets sensibilisés à la digitaline, bactéries nouvelles capables de transformer les sels de bismuth en dérivés toxiques – qui sont en cause. Peut-être n’est-il pas inutile à ce propos de rappeler que la pharmacologie est une science biologique et que bon nombre de réactions imprévues aux médicaments ne s’expliquent que par la prodigieuse diversité de récepteurs qui caractérise l’espèce humaine. Ces quelques exemples montrent l’intérêt de la pharmacovigilance:– il est indispensable de recueillir les effets adverses des médicaments, c’est-à-dire de surveiller de façon permanente la population traitée;– les données obtenues n’ont qu’une valeur instantanée, elles doivent donc être systématiquement complétées;– du point de vue de la pharmacovigilance, l’étude d’un médicament n’est jamais terminée.Objectifs de la pharmacovigilanceCe sont d’abord la surveillance du prévisible et la recherche de l’imprévisible en matière d’effets médicamenteux. En d’autres termes, il s’agit à ce stade de surveiller de façon régulière l’importance des effets adverses déjà connus et, d’autre part, de déceler ceux qui n’avaient jamais été observés auparavant. À ce double objectif s’attache souvent une notion d’urgence: les accidents graves doivent être connus puis corrélés à la prise médicamenteuse le plus vite possible, ce qui suppose à la fois un véritable quadrillage de la population traitée et des moyens d’information rapides. De façon moins urgente et à plus long terme, l’objectif de la pharmacovigilance est de connaître, dès qu’elles deviennent significatives, toute variation de la fréquence d’un effet indésirable et toute apparition d’un nouvel effet adverse. La quantification des données utilise les méthodes épidémiologiques; c’est ainsi que l’on définit:– l’incidence d’un effet adverse, nombre de nouveaux cas observés pendant une certaine période;– la prévalence d’un effet adverse, nombre total de cas présents à un certain moment ou pendant une certaine période: c’est la somme des cas anciens persistants et des nouveaux cas.Ces valeurs, lorsqu’elles sont rapportées à l’ensemble de la population traitée, permettent de distinguer respectivement la fréquence des nouveaux cas et la fréquence instantanée d’un effet adverse. À titre d’exemple, le risque de survenue de quelques cytopénies médicamenteuses, évalué en Suède sur une période de cinq ans (1966-1970), se décompose comme le montre le tableau 1. Plus récemment, une augmentation importante des cas d’agranulocytose liés à l’administration d’un neuroleptique, la clozapine, a été constatée en Finlande (tabl. 2).Il est clair que de tous les pays où ce médicament est commercialisé, la Finlande a été la seule à présenter cette brutale augmentation de fréquence qui semble d’ailleurs s’être déclarée très rapidement, en quelques mois. Un facteur local de majoration de cet effet toxique bien connu est pratiquement certain; il a entraîné le retrait rapide (juill. 1975) de la clozapine du marché finlandais. Une telle décision ne se justifiait pas dans les autres pays où ce médicament est toujours utilisé sans que l’on ait observé jusqu’à présent d’augmentation de la fréquence des cas d’agranulocytose. Le mécanisme de survenue de cet effet n’est pas encore connu à l’heure présente.Si le premier objectif de la pharmacovigilance est de recueillir un maximum de données, le second est évidemment de pouvoir les redistribuer. Le retour au praticien est une étape fondamentale qui selon la gravité des cas peut revêtir deux formes principales:– l’alerte, dans toutes les éventualités d’effets toxiques majeurs, peut être donnée rapidement à chaque médecin et pharmacien, par l’intermédiaire de leurs ordres respectifs;– l’information régulière, qui doit être considérée comme une partie indispensable de l’enseignement postuniversitaire, n’est pas vraiment systématique, mais de nombreuses initiatives existent dans ce domaine.Enfin, les résultats obtenus servent de banque de données tant pour les pouvoirs publics que pour l’industrie pharmaceutique. Il y a là un système de référence dont la comparaison aux médicaments en expérimentation permettra de mieux préciser l’importance de leurs effets adverses et ainsi d’évaluer de façon plus rigoureuse un des aspects de leur intérêt thérapeutique.Les structures internationalesLe Centre international de pharmacovigilance de l’O.M.S. ou WHO drug monitoring center. Décidé à titre expérimental en 1963, le Centre mondial est définitivement créé en 1971 et Genève choisie comme lieu d’implantation. Les activités de ce centre sont multiples: centraliser les rapports que lui adressent les centres nationaux (Allemagne fédérale, Australie, Canada, États-Unis, France, Grande-Bretagne, Irlande, Nouvelle-Zélande, Israël, Japon, Pologne, Pays-Bas, Suède, Danemark, Norvège); les informations sont traitées à l’aide de systèmes informatisés, et une analyse mensuelle est adressée aux participants: harmoniser la terminologie, standardiser les critères d’évaluation des réactions indésirables, recevoir communication des décisions officielles prises par chaque pays membre concernant les médicaments dans le cadre de la pharmacovigilance et les transmettre aux autres pays; susciter des enquêtes au niveau des centres nationaux, qu’elles soient monocentriques ou multicentriques; provoquer des réunions d’experts et la publication de leurs travaux, en particulier en ce qui concerne la méthodologie. Les informations répercutées par ce centre international ne sont transmises qu’aux gouvernements des États participants, et ce confidentiellement, chaque gouvernement étant libre de les utiliser comme bon lui semble.Les centres nationaux de pharmacovigilance. Créés à l’instigation de l’O.M.S., ils se rapprochent plus ou moins de la structure type qu’elle préconise: une équipe permanente technique d’exécution, souvent composée de fonctionnaires de l’État aidés par des spécialistes scientifiques et médicaux, pharmaciens, épidémiologistes, statisticiens, informaticiens..., un comité national composé d’experts en médecine clinique, en pharmacologie, en toxicologie, en épidémiologie et en statistiques. Le rôle de chaque centre national devrait être triple: recueillir et évaluer les effets indésirables, organiser des enquêtes, enfin assurer l’information et le conseil thérapeutique.Les structures françaisesL’organisation de la pharmacovigilance en France remonte à l’arrêté promulgué le 2 décembre 1976 (Journal officiel du 19 décembre 1976) par le ministère de la Santé.Il avait pour objet la mise en place de trois structures de contrôle agissant en coordination. Des centres de pharmacovigilance hospitalière étaient institués dans les centres hospitaliers régionaux. Un Centre national de pharmacovigilance (C.N.P.V.) devait collationner les informations provenant de tous les secteurs de l’activité sanitaire. Auprès du ministre de la Santé était constituée une commission technique qui avait pour mission de proposer au ministre les décisions à prendre à la suite des constatations faites par les instances ayant recueilli les données.À ces structures, le décret du 24 mai 1986 a ajouté une quatrième instance, renforçant ainsi le dispositif comme indiqué ci-dessous.Le Centre national de pharmacovigilance a été créé en 1973, fondé par les membres du Conseil de l’ordre des médecins, le Conseil de l’ordre des pharmaciens, les centres anti-poisons, le Syndicat national de l’industrie pharmaceutique; il travaillait avec les centres hospitaliers de pharmacovigilance, son rôle a un peu changé depuis 1984.Les centres régionaux de pharmacovigilance tous placés, à la demande du ministère de la Santé, dans des centres hospitaliers et universitaires (C.H.U.) auprès d’un service de pharmacologie ou de centres de traitement des intoxications. Les centres régionaux de pharmacovigilance sont actuellement au nombre de 30. Le décret de 1984 demande aux médecins de déclarer obligatoirement les effets inattendus ou toxiques (effets indésirables, mauvais usages...) des médicaments qu’ils ont prescrits au centre de pharmacovigilance le plus proche. Les centres conduisent les enquêtes demandées par le ministre et enregistrent les données dans un système informatique commun. Ils ont également pour mission de diffuser l’information sur les médicaments.Le Comité technique de pharmacovigilance coordonne et évalue les enquêtes effectuées par les centres régionaux.La Commission nationale de pharmacovigilance apprécie les résultats de ces enquêtes et donne des avis au ministère de la Santé dont elle dépend directement sur les mesures à prendre pour protéger la santé publique (information, modification des conditions d’utilisation, retrait éventuel du marché).Parallèlement, les industriels titulaires d’une autorisation de mise sur le marché (A.M.M.) d’une spécialité pharmaceutique sont tenus de déclarer tous les effets inattendus ou toxiques de leur spécialité directement à la Direction de la pharmacie et du médicament qui assure le secrétariat du Comité technique et de la Commission nationale de pharmacovigilance. Les mesures décidées par le ministre chargé de la santé sont divulguées par voie de presse si elles sont importantes. Cette déclaration doit se faire tous les semestres la première année de la commercialisation ou annuellement ensuite. Les modifications se répercutent dans l’A.M.M. et la monographie de la spécialité concernée.La notification des effets adversesLa notification est assurée essentiellement par les médecins ou les pharmaciens, qui ont à leur disposition une fiche de recueil des données, analogue à celle qu’utilisent les médecins praticiens, et qui peuvent aussi faire parvenir ces notifications à la Commission nationale via l’ordre des pharmaciens. Les professions paramédicales citées dans l’article 3 de l’arrêté ont aussi la possibilité d’en utiliser.Dans le cadre hospitalier, le problème qui se pose actuellement est de savoir comment améliorer la notification des réactions adverses par tous les hôpitaux dans le cadre des centres hospitaliers de pharmacovigilance déjà existants, qu’il s’agisse de la surveillance systématique ou de la mise en garde immédiate.À qui incombe la notification d’un effet adverse observé au lit d’un malade? En premier lieu, elle revient au(x) médecin(s) responsable(s) du lit. Cette activité supplémentaire peut, à première vue, rebuter un médecin déjà très occupé. Plusieurs solutions sont possibles, dont la plus commode serait peut-être la désignation d’une personne du service (médecin, étudiant médecin, infirmière), responsable de prendre systématiquement contact avec le centre de pharmacovigilance local. Il nous semble également que les pharmaciens de chaque établissement hospitalier pourraient utilement seconder les cliniciens dans ce travail de notification. Il existe actuellement une fiche cartonnée, dite fiche d’alerte, très simple et très rapide à remplir, qui suffit à jouer son rôle de pense-bête auprès du médecin et de matériel de notification à adresser au centre local. Ce rapport très succinct sert de base pour l’établissement d’une fiche beaucoup plus détaillée, par un des membres du centre. Cette fiche, dite fiche en clair, est codée sur un document et envoyée au centre informatique du Comité technique de pharmacovigilance qui regroupe les données pour l’ensemble de la France.Tout effet secondaire, du plus fréquent au plus inattendu, doit être notifié. Il n’y a pas de critère de présélection dans les notifications à faire. Certes, il peut paraître inutile parce que très connu de signaler une atteinte gastrique avec de l’aspirine. Il se peut cependant, chez un sujet donné, qu’on s’aperçoive en étudiant l’anamnèse que l’aspirine était bien tolérée avant l’intervention de tel ou tel facteur concomitant. Ainsi, d’un faisceau d’observations peut se dégager un certain nombre de conditions qui pondèrent la responsabilité d’un médicament dans le déclenchement d’une pathologie iatrogène. Quand l’alerte est ainsi donnée, les cliniciens hospitaliers sont bien placés pour apporter une contribution précieuse à la pharmacovigilance en aidant à faire l’examen critique de la relation envisagée entre le médicament et la réaction adverse.Il est possible de procéder de deux manières pour recueillir les effets adverses. La première consiste, au cours des traitements, à noter tout effet adverse au fur et à mesure qu’il est constaté: c’est la pharmacovigilance extensive ou spontanée , dont la méthodologie s’apparente à l’épidémiologie descriptive. Dans ce genre de pharmacovigilance, tous les praticiens de l’aire géographique du centre sont concernés et la bonne marche du travail dépend d’abord d’eux. La qualité des notifications adressées au centre dépend en effet de leur bonne foi: les informations tronquées, dissimulées, ou omises involontairement faussent en effet le système dès le départ. Elle dépend aussi de leur compétence à identifier l’effet adverse, de l’intérêt qu’ils prennent à la question, du temps dont ils disposent. Discipline médicale nouvelle, presque encore au stade expérimental, du moins en France, elle ne fait pas encore l’objet d’un enseignement systématique dans les facultés de médecine. Ce système de recueil des données est limité par plusieurs facteurs. Les données recueillies par la notification spontanée restent numériquement trop faibles par rapport aux événements réels pour constituer un échantillon statistiquement valable; de plus, le nombre d’administrations du médicament (importance de sa consommation) n’est au mieux qu’approximativement connu. Il n’est donc pas possible de calculer une fréquence exacte de survenue, mais tout au plus de se faire une idée de l’importance du phénomène. De plus, la pharmacovigilance extensive n’apporte pas d’explication du mécanisme des accidents. Au total, la pharmacovigilance extensive permet de soupçonner la relation de cause à effet médicament-effet adverse, de regrouper des facteurs de corrélation éventuels: âge, sexe, ... de signaler des interactions médicamenteuses, et elle aboutit à formuler des hypothèses qui, si elles sont suffisamment argumentées et les faits envisagés suffisamment graves, peuvent justifier la prise de mesures d’urgence comme ce fut le cas en France avec les encéphalopathies mortelles après l’utilisation de sous-nitrates de bismuth.La seconde méthode de travail s’appelle la pharmacovigilance intensive , dont un des objectifs est de confirmer les données de la pharmacovigilance extensive. Elle étudie les problèmes de pharmacovigilance dans des limites définies de temps et/ou d’espace. Elle fait appel à des techniques de recueil systématique (et non plus spontané) des données. Elle peut se faire par malade, par médicament, par effet indésirable et, dans chacun de ces cadres, elle consiste à relever respectivement tous les effets adverses chez un groupe de malades, tous les effets adverses d’un certain type chez tous les malades quel que soit le médicament.Dans les méthodes dites numériques, on procède par des études prospectives directement sur une population, ou par des études rétrospectives, sur dossier, moins satisfaisantes, car elles comportent plus de biais importants. Dans les méthodes par estimations, on procède à l’étude des registres de mortalité et à l’estimation du risque ajouté par un médicament à celui qui existe déjà spontanément de voir survenir une pathologie donnée (par exemple, relation entre œstroprogestatifs et thrombophlébites).Dans les études numériques, on peut calculer avec précision une fréquence de survenue d’un effet et s’assurer de sa spécificité en utilisant une population témoin; dans les études par estimations, on isole le facteur par lequel la prise d’un médicament multiplie le risque de survenue de l’effet indésirable.Traitement de la notificationLe traitement de la notification comporte plusieurs étapes. D’abord, il faut préciser si la notification correspond bien à un effet indésirable, et, si oui, s’il est dû à la prise d’un ou de plusieurs médicaments (spécificité), établir sa gravité et sa fréquence, déterminer son mécanisme. Établir l’imputabilité d’un effet indésirable reconnu comme tel à l’action d’un médicament est une opération complexe, car elle requiert que soient réunis un certain nombre de critères pour qu’on puisse situer la responsabilité du médicament dans une appréciation globale dont les termes sont: certain, probable, possible, éventuel et enfin douteux. Chacun de ces termes correspond à la présence d’un certain nombre de critères d’imputabilité qui servent à l’étude analytique de la réaction indésirable et qui sont la chronologie de l’événement, la disparition des troubles lors de la réinduction du traitement, l’absence d’autres causes pouvant expliquer l’effet, l’existence d’examens complémentaires biologiques déterminants (dosage du médicament, par exemple), le caractère connu et non connu de l’effet indésirable (bibliographie). De plus, chacun des critères est évalué de 0 à 3: 0 = non recherché, 1 = négatif ou inconnu, 2 = intermédiaire ou douteux, 3 = positif ou évocateur. Le problème de l’imputabilité est compliqué par le fait des associations médicamenteuses soit à l’intérieur de la même forme galénique, soit par la polymédication. À partir du moment où l’effet adverse est identifié comme tel, et évalué dans son rapport avec le traitement médicamenteux, sa fréquence, sa gravité, il est transmis, en France, par le centre de pharmacovigilance référencé (c’est-à-dire local) qui en a fait l’étude à un centre de traitement informatisé des données de pharmacovigilance créé et situé à Lyon, qui permet une mise en mémoire de toutes les notifications recueillies en France. Cette transmission se fait à l’aide du dossier codé à partir de l’observation prise en langage clair. Le codage est réalisé par les équipes des centres à l’aide de dictionnaires qui permettent une standardisation des entrées des données et qui sont un dictionnaire des médicaments et un dictionnaire des symptômes et des réactions adverses. L’accès à ces données informatisées est régi par une convention tripartite: ministère de la Santé, Association des unités de pharmacovigilance hospitalière, structure informatique lyonnaise. Cette convention garantit l’accès limité à un personnel de pharmacovigilance et officiellement reconnu. Ce centre informatique français transmet ses données à celui de l’O.M.S., dont l’ordinateur garde en mémoire l’ensemble des informations et adresse tous les trois mois aux pays participants un rapport qui comporte: toutes les réactions notifiées, classées par noms de médicaments; tous les médicaments mentionnés, classés par effets adverses présumés; tous les effets adverses signalés pour la première fois; la liste des réactions létales et les malformations congénitales, des «signaux» attirant l’attention sur les augmentations au-dessus d’un certain seuil de fréquence au cours de la dernière période; des rapports concernant un effet indésirable ou un médicament relativement à leur fréquence dans la mémoire générale. Chaque rapport porte sur les derniers trois mois. À l’heure actuelle, les notifications faites par le secteur médical non hospitalier sont de plus en plus fréquemment adressées aux centres hospitaliers de pharmacovigilance qui les regoupent. Les réflexions suggérées par les problèmes de recueil et de traitement des données par les centres s’appliquent donc également à la pharmacovigilance non hospitalière.
où Ct représente la concentration plasmatique à l’instant t , A et 見 les caractéristiques du premier compartiment, dit compartiment central, B et 廓 celles du compartiment suivant, N et n les caractéristiques du n -ième compartiment.L’analyse compartimentale attribue à chaque exponentielle la valeur d’un compartiment particulier, celui-ci étant défini de façon dynamique comme la masse de substance ayant, au même moment, un comportement identique dans l’organisme (fig. 2). Ainsi, selon la nature de la courbe de décroissance des concentrations plasmatiques et le nombre de processus exponentiels auxquels elle correspond, on distingue des cinétiques monocompartimentales, bicompartimentales ou multicompartimentales.Dans chaque compartiment, le devenir du médicament est caractérisé par son volume de distribution et sa demi-vie .Le premier est un volume apparent, théorique, sans correspondance anatomique ou physiologique. Il indique simplement le volume dans lequel la masse de médicament serait dissoute en l’absence de toute fixation: il permet d’avoir une idée de l’importance de la diffusion et de la fixation du médicament. La demi-vie correspondante indique le temps au bout duquel la concentration est devenue la moitié de la concentration initiale.Dans le cas d’un modèle à plusieurs compartiments, il est possible de calculer les constantes de transfert qui caractérisent l’intensité des flux de médicament entrant et sortant d’un compartiment donné. Un des cas les plus fréquemment rencontrés est celui d’un modèle ouvert à deux compartiments (fig. 2). Le compartiment central est celui où le médicament est injecté et d’où il est éliminé. Le compartiment périphérique (ou profond) représente les localisations tissulaires du médicament; son volume est plus grand, ses échanges avec le premier compartiment sont lents.Lorsque le médicament n’est pas administré par voie intraveineuse mais sous une forme qui doit être résorbée, on peut mesurer la constante de résorption correspondante. Celle-ci a l’intérêt de permettre l’appréciation de la durée de la résorption de la dose administrée. Lorsque celle-ci est lente, un effet retardé et prolongé est vraisemblable. En cas de résorption digestive, une vitesse trop lente peut cependant entraîner une résorption partielle (cf. infra , Biodisponibilité des formes pharmaceutiques ).Des différentes données pharmacocinétiques caractérisant un médicament, il est possible de tirer des renseignements importants concernant son utilisation chez l’homme. La notion d’un compartiment périphérique de grand volume est intéressante lorsqu’il s’agit par exemple de sélectionner un antibiotique destiné à traiter une localisation infectieuse profonde. Au contraire, la recherche d’un effet vasculaire orientera le choix du thérapeute vers des médicaments de faible volume de distribution.Avec la demi-vie d’élimination d’un médicament mesuré pour un individu déterminé, il est possible de calculer avec précision le rythme de son administration de manière à maintenir une imprégnation continue de l’organisme.On conçoit enfin que foie et reins étant les principaux organes d’élimination des médicaments, leur efficacité conditionne l’activité médicamenteuse. Toute atteinte fonctionnelle hépatique ou rénale peut ainsi modifier la cinétique et les caractéristiques des effets pharmacologiques attendus: il est possible de pallier cet inconvénient en connaissant la part respective de ces deux émonctoires dans le métabolisme des médicaments. Ainsi, dans l’insuffisance hépatique grave, on choisira de préférence des médicaments éliminés par les reins. En cas d’insuffisance rénale, on retiendra au contraire les substances dégradées par le foie.3. Biodisponibilité des formes pharmaceutiquesDiversité des formes pharmaceutiquesLe médicament, sous sa forme initiale purifiée, n’est pas directement utilisable en thérapeutique. En effet, il s’agit le plus souvent d’une poudre (digoxine, sulfate d’atropine) ou d’un liquide (clofibrate) dont l’administration en faible quantité est impossible ou très peu commode. On est donc obligé de l’administrer sous une forme pharmaceutique déterminée où l’on retrouvera l’exacte quantité que l’on souhaite délivrer au malade. Cette méthode suppose que la dose apportée soit aussi la dose active dans l’organisme. Or ce n’est pas toujours le cas, et différents travaux ont pu montrer qu’un même médicament, administré au même individu, à la même posologie, mais sous des formes pharmaceutiques différentes, pouvait développer des effets d’intensité variable. En d’autres termes, malgré des sujets et des posologies identiques, des formes pharmaceutiques différentes ne développaient pas le même effet. Cette observation a été faite en particulier avec des comprimés de digoxine de marques différentes, avec des gélules de chloramphénicol et de tétracycline. Elle s’explique par une inadéquation de la forme pharmaceutique, qui a deux causes principales. La première réside dans le fait que celle-ci peut ne libérer qu’une fraction du médicament qu’elle contient: la quantité résorbée est donc inférieure à la dose administrée (cf. chap. 1 et 2), la fraction restante étant éliminée dans les selles. Dans d’autres cas, la forme pharmaceutique libère bien la totalité du médicament qu’elle contient, mais avec une vitesse si lente que les concentrations plasmatiques correspondantes (après résorption) seront toujours faibles. Le gradient de concentration entre le plasma et les tissus le sera également: la diffusion tissulaire sera peu importante, l’effet pharmacologique résultant sera faible, voire nul.On comprend ainsi qu’après avoir sélectionné un médicament il est absolument indispensable de l’administrer sous une forme pharmaceutique telle qu’il puisse, à partir d’elle, développer la totalité des effets que l’on attend de lui. C’est de cette constatation qu’est née la biopharmacie , discipline qui étudie les relations qui existent entre les propriétés physico-chimiques du médicament et de ses formes pharmaceutiques et les effets pharmacologiques qu’ils produisent. La notion de biodisponibilité ou disponibilité biologique vise à corréler la dose administrée à l’effet qu’elle produit lorsqu’elle est utilisée sous une forme pharmaceutique résorbable. La notion de bioéquivalence envisage l’efficacité relative d’un même médicament, administré à la même posologie, sous des formes pharmaceutiques différentes. Les formes pharmaceutiques sont bioéquivalentes si elles produisent des effets d’intensité et de durée identiques; dans le cas contraire, elles ne le sont pas et alors il est important de ne pas remplacer l’une par l’autre.Paramètres de biodisponibilitéThéoriquement, la biodisponibilité d’une forme pharmaceutique devrait être définie par deux paramètres: la quantité de médicament mise à la disposition des sites d’action et la vitesse de cette mise à disposition. Or, cette définition théorique est peu réaliste. En effet, quantifier ces deux paramètres au niveau du site d’action est difficilement réalisable chez l’homme. En revanche, la détermination de ces paramètres peut se faire plus aisément si l’on considère non plus les sites d’action, mais la quantité de médicament présente dans la circulation sanguine. On mesure alors une biodisponibilité systémique . De nombreux travaux ont en effet montré que, pour un grand nombre de médicaments, il existe une relation entre leurs concentrations sanguines et leurs effets pharmacologiques tissulaires; la concentration sanguine, en d’autre termes, est dans une certaine mesure le reflet de la concentration au niveau du site d’action induisant l’effet pharmacologique. D’où la définition suivante pratiquement admise par tous: la biodisponibilité d’une forme pharmaceutique représente la fraction de médicament administré qui atteint la circulation générale et la vitesse avec laquelle elle l’atteint. Par définition, le médicament contenu dans une forme pharmaceutique administrée par voie intraveineuse a une biodisponibilité de 100 p. 100, puisque, dans ces conditions, on évite l’étape de la résorption qui est donc à la fois totale et instantanée. Administré par une autre voie (orale, rectale, sublinguale, etc.), le médicament va devoir traverser un certain nombre de barrières biologiques qui peuvent représenter une limitation de son passage dans la circulation sanguine. De ce fait, la biodisponibilité s’exprimera alors par comparaisons. On distingue ainsi:– la biodisponibilité absolue. Elle est évaluée par comparaison avec la voie intraveineuse. Par exemple, on évalue la biodisponibilité absolue d’un comprimé de digoxine en comparant les concentrations sanguines de digoxine obtenues au cours du temps après administration orale avec celles obtenues après injection intraveineuse de la même dose;– la biodisponibilité relative. Dans ce cas, on administre une forme pharmaceutique de biodisponibilité connue, dite forme de référence, par une voie autre que la voie intraveineuse. Lorsqu’on compare la biodisponibilité de différents comprimés contenant le même médicament, dont la mise en forme pharmaceutique a été différente, celui d’entre eux présentant la meilleure biodisponibilité relative exprimera la biodisponibilité relative optimale.Facteurs de biodisponibilitéLes facteurs influençant la biodisponibilité d’une forme pharmaceutique sont multiples. Les caractères physico-chimiques de la molécule peuvent être d’abord incriminés: ils conditionnent sa solubilité et donc sa vitesse de dissolution ainsi que sa résorption. Or, pour être résorbé, un médicament doit d’abord être solubilisé (fig. 3). Sa vitesse de dissolution dépend de ses formes cristallines et/ou chimiques (estérification, salification, état d’hydratation); elle est généralement fonction de la taille des particules et de l’état d’ionisation de la molécule. De même, pour la fabrication de la forme pharmaceutique, le médicament doit être mélangé à divers excipients, c’est-à-dire à des substances inactives par elles-mêmes qui facilitent son administration et sa conservation. Certains excipients ont tendance à absorber une fraction du médicament de sorte que la quantité parvenant dans la circulation générale est alors moindre. D’autres modifient la vitesse de dissolution du médicament, réduisant ou augmentant ainsi sa biodisponibilité. Les conditions de fabrication peuvent également influencer la biodisponibilité: lors de la fabrication d’un comprimé, la compression du mélange à mettre en forme, pouvant légèrement varier d’un jour à l’autre, modifiera la vitesse de dissolution. Enfin, les conditions de conservation de la forme pharmaceutique peuvent être responsables de la dégradation chimique ou d’une modification cristalline et entraîner ainsi une évolution de la biodisponibilité.Mesure de la biodisponibilitéLa biodisponibilité d’une forme pharmaceutique peut être déterminée soit indirectement, par la mesure de l’effet qu’elle produit, soit directement, en mesurant la quantité contenue dans l’organisme.La première méthode est intéressante dans la mesure où elle permet de mesurer directement la bioéquivalence des deux formes pharmaceutiques d’un même médicament. Elle se heurte malheureusement à la difficulté de mesurer avec précision l’intensité de certains effets thérapeutiques et, de ce fait, n’est que rarement employée.Le deuxième groupe de méthodes fait appel à l’analyse pharmacocinétique du médicament dans les différents liquides biologiques (sang et/ou urines) après administration unique ou répétée. La détermination de la quantité de médicament résorbée par la mesure des taux sanguins repose sur la loi de Dost (1964) ou principe des aires correspondantes: «Quelle que soit la voie d’administration et quel que soit le nombre de compartiments de distribution dans l’organisme, la quantité résorbée est toujours proportionnelle à l’aire sous la courbe des concentrations sanguines extrapolée de 0 à l’infini.» Pour être utilisable, ce principe impose trois contraintes principales:– il doit exister dans l’organisme un compartiment central incluant le sang à partir duquel se produit l’élimination ; l’expérience a montré que ce postulat est vérifié dans la très grande majorité des cas;– la cinétique de l’élimination du médicament doit être de premier ordre; cette condition est souvent réalisée, mais ce n’est pas toujours le cas, et il faut toujours la vérifier;– le volume du compartiment central et la constante de vitesse globale d’élimination (ke ) restent constants au cours des essais; ce dernier point n’est jamais rempli.Il est donc nécessaire de mesurer ces paramètres et de corriger l’aire sous la courbe en fonction de leurs valeurs respectives au moment de l’étude. À partir de ce principe, si ces conditions sont remplies, il est donc possible de mesurer la biodisponibilité absolue ou relative d’une forme pharmaceutique par comparaison des surfaces sous la courbe correspondant aux différentes formes. Cette surface peut être calculée après administration unique ou lors d’une administration chronique, entre deux administrations. Parallèlement à la quantité globale de médicament résorbé, on peut également mesurer la vitesse de résorption à partir des taux plasmatiques. Il est en effet possible, à partir d’une étude pharmacocinétique des taux plasmatiques observés, de calculer la constante de résorption selon un modèle compartimental. Mais il est aussi possible de mesurer simplement la concentration plasmatique maximale et le temps nécessaire pour l’obtenir après l’administration. Ces deux paramètres sont en effet directement liés à la vitesse de résorption; lorsqu’elle diminue, la concentration plasmatique maximale diminue et le temps nécessaire pour y parvenir augmente, et réciproquement. Les mêmes renseignements peuvent être retrouvés à partir des concentrations urinaires. En effet, si un médicament est éliminé uniquement dans les urines et sans transformation, la quantité éliminée est alors égale à la quantité résorbée. On peut ainsi dans ce cas particulier évaluer la biodisponibilité absolue sans recourir à l’administration intraveineuse. Si le médicament est métabolisé, il en sera de même si l’on connaît tous les métabolites et que l’on est capable de mesurer leur excrétion. L’analyse pharmacocinétique des données urinaires peut également fournir des renseignements concernant la vitesse de résorption du médicament, mais cette analyse est généralement plus difficile à réaliser et moins précise que l’étude des concentrations plasmatiques.Enfin, de nombreuses études ont permis de corréler la biodisponibilité d’une forme pharmaceutique in vivo chez l’homme avec des tests de dissolution in vitro . Il n’a pas été possible à ce jour de dégager une relation générale, mais ce type d’essai in vitro peut être utilisé pour vérifier l’égalité de biodisponibilité entre deux lots de fabrication, à la condition que le test in vitro ait été étalonné au préalable vis-à-vis de la biodisponibilité in vivo .Problèmes pratiquesIl est nécessaire de définir les critères de jugement concernant la biodisponibilité d’une ou de plusieurs formes pharmaceutiques. Deux différentes approches peuvent être utilisées. D’un point de vue mathématique, l’analyse statistique à l’aide de tests paramétriques ou non paramétriques permet avec un risque d’erreur connu de décider si deux formes pharmaceutiques sont différentes ou non et de calculer un intervalle de confiance correspondant. Mais, d’un point de vue clinique, il est également nécessaire de considérer la variation d’effets qui peuvent être engendrés. Ainsi, on s’accorde généralement pour considérer que deux médicaments sont bioéquivalents si leur biodisponibilité ne diffère pas de plus de 20 p. 100. Mais ce critère ne peut être le seul à considérer. Si, en effet, on se trouve en présence de deux formes, l’une ayant une biodisponibilité de 50 p. 100 avec une variabilité de 10 p. 100, l’autre de 75 p. 100 avec une variabilité de 25 p. 100, il sera préférable d’utiliser la première forme, bien que sa biodisponibilité soit plus faible. En effet, la réponse thérapeutique sera alors beaucoup plus reproductible et cette forme pharmaceutique plus maniable.Les études de biodisponibilité, requises pour commercialiser une nouvelle forme pharmaceutique (étude in vivo ) et pour s’assurer de l’adéquation de tout nouveau lot de fabrication (étude in vitro ), permettent de vérifier et de garantir la bioéquivalence des préparations d’un même médicament mises à la disposition du praticien. Si elles apportent une grande sécurité sur le plan de la fiabilité de ces formes pharmaceutiques, elles ne peuvent cependant garantir de façon absolue l’efficacité des médicaments qu’elles contiennent. Tous les autres facteurs propres au sujet traité demeurent: ainsi, tout trouble pathologique de la résorption intestinale peut diminuer la quantité de médicament résorbée, même si la forme pharmaceutique utilisée a une excellente biodisponibilité. Plus simplement, l’apport simultané de plusieurs médicaments, la prise d’aliments peuvent modifier quantité résorbée et vitesse de résorption de l’un ou de plusieurs d’entre eux. L’étude de biodisponibilité n’est plus en cause dans ces cas: elle apporte seulement, et c’est déjà un intérêt considérable, la preuve que les formes pharmaceutiques sont adéquates. La modification pharmacologique observée relève alors d’une autre cause, d’ordre individuel, qu’il convient de déterminer.4. Essais thérapeutiquesQuelle que soit la qualité des travaux effectués lors de l’étude préclinique d’un nouveau médicament, les résultats obtenus sur des animaux n’ont qu’une valeur d’orientation prévisionnelle des effets que l’on pourra observer chez l’homme. La première fois qu’un nouveau médicament est administré à un homme, il s’agit donc bien d’une expérimentation. Cela explique les réticences d’ordre éthique qui sont ressenties par tous médecins, légistes, moralistes, patients...Cependant, les progrès réalisés au cours des dernières décennies dans le traitement de nombreuses maladies sont une raison suffisante et une justification à persévérer dans la voie des essais puisque ces traitements nécessitent soit une amélioration des médicaments utilisés pour les traiter, soit la découverte de nouveaux produits efficaces. Les recherches se poursuivant activement aux niveaux chimique et pharmacologique préclinique, il y aura donc toujours dans l’histoire naturelle de ces nouveaux médicaments, actuels ou futurs, un premier passage sur l’homme.Les règles de l’éthique médicale imposent de ne faire ces essais que dans des conditions strictement définies: les risques, inhérents à toute nouvelle thérapeutique, doivent être largement compensés par les bénéfices escomptés. Les essais doivent être parfaitement codifiés afin que les résultats obtenus puissent être interprétés sans équivoque. Toutes les garanties de surveillance par des équipes médicales, compétentes et entraînées, doivent être données aux sujets qui recevront le nouveau médicament.Phases de l’essai clinique du médicamentLa nécessité d’une expérimentation parfaitement planifiée, garantissant le minimum de risques aux sujets recevant le nouveau médicament, s’est fait jour dans l’esprit des cliniciens il y a plusieurs décennies, mais ce n’est que depuis peu que l’investigation clinique d’un nouveau médicament a été divisée en plusieurs phases. Codifiées à partir de données des études de pharmacologie préclinique ou animale, les phases, au nombre de quatre dans la classification de la F.D.A. (Food and Drug Administration), sont telles que la progression d’une phase à l’autre est conditionnée par les résultats obtenus à chaque étape.Phase I, ou première administration à l’hommeAu cours de cette phase, sont étudiés l’activité biologique générale, la pharmacocinétique, le métabolisme et la tolérance du nouveau produit. Ces divers éléments sont étudiés sur un petit nombre de sujets volontaires sains. Le problème juridique du volontaire sain recevant un nouveau médicament est résolu dans la majorité des pays, notamment ceux de l’Union européenne. À la suite de la phase I qui dure de un an à un an et demi, la tolérance de l’organisme au produit nouveau étant définie, la connaissance de ses caractéristiques pharmacocinétiques permet de proposer une posologie , c’est-à-dire une quantité de médicament qui, administrée selon un certain rythme, aura un maximum de chances d’entraîner un effet thérapeutique, sans effet toxique. C’est également au cours de cette phase I que certaines interactions peuvent être étudiées: par exemple l’interaction de l’alcool avec les anxiolytiques.Phase IIAu cours de la phase I, l’activité thérapeutique , c’est-à-dire la possibilité de traiter une maladie ou un symptôme, n’a pas du tout été abordée. Pendant la phase II, qu’on subdivise parfois assez arbitrairement en phase II précoce et en phase II tardive, auront lieu des essais cliniques , sur des malades sélectionnés, pendant une période allant de un à deux ans. Les études de la phase II s’appuient sur les résultats obtenus en phase I. Elles vont permettre de définir:– la validité des posologies proposées;– l’existence d’une relation entre le médicament et les effets thérapeutiques: relation dose-effet ou relation concentration plasmatique-effet.S’adressant à des sujets malades, on étudie également au cours de cette phase l’influence de la maladie sur le métabolisme et la pharmacocinétique du nouveau médicament. On peut aussi observer dès cette phase l’existence d’interactions médicamenteuses. Les recherches effectuées en phase II le sont dans des services hospitaliers hautement spécialisés. Les réactions des malades sont suivies de très près par des médecins spécialistes, ce qui assure aux malades les garanties le plus complètes possible de sécurité.Phase IIILes résultats des phases I et II permettent de décider de la poursuite ou de l’arrêt de l’étude du nouveau produit. En cas de poursuite, la phase III permet, en se plaçant dans les conditions le plus proches possible de la pratique médicale quotidienne, de confirmer les informations obtenues au cours des phases précédentes et de définir les conditions exactes de l’utilisation du nouveau produit. On définit ainsi les indications et les contre-indications d’utilisation dans l’arsenal thérapeutique disponible. Cette phase est, par ses techniques et méthodologies, proche de la phase II. Elle en diffère cependant par l’utilisation de grandes séries de malades traités (essais contrôlés), ce qui nécessite une très grande rigueur expérimentale, afin de permettre un traitement statistique correct des informations. Elle ne nécessite pas, par contre, de services médicaux hautement spécialisés puisque, en phase II, les inconvénients majeurs qui pourraient être liés au nouveau médicament ont été détectés. Elle aboutit à l’autorisation de mise sur le marché (A.M.M.).Phase IVElle suit la mise du nouveau médicament sur le marché. Elle n’appartient plus tout à fait à une phase d’«essais» cliniques, mais elle est pourtant indispensable pour mettre en évidence les effets médicamenteux indésirables qui, compte tenu de la durée relativement courte des phases I, II et III, n’auraient pas pu apparaître.C’est également durant cette phase que seront complétées les informations concernant des médicaments qui sont administrés au long cours, pour des maladies chroniques de longue durée. Il faut également rappeler que c’est à partir d’observations effectuées par des cliniciens curieux, en phase IV, que de nouvelles indications thérapeutiques peuvent apparaître. À ce stade, on recherche, après les effets indésirables, les indications nouvelles, interactions particulières. Enfin, cette phase est celle au cours de laquelle les rapports risque-bénéfice et coût-bénéfice pourront être établis.Techniques d’études utilisées lors des essaisIl existe deux grands types de méthodes: les essais dits ouverts, les essais dits contrôlés. Nous allons rapidement les passer en revue en essayant de dégager les avantages et les inconvénients de chacune de ces techniques et en précisant leur place dans les différentes phases. Quelle que soit la méthode utilisée, elle nécessite une équipe médicale parfaitement entraînée et attentive, tout au long de l’essai, à suivre les étapes du plan expérimental établi.Ce plan expérimental devra, lui, être soumis à des règles strictes d’éthique qui garantiront dans tous les cas une parfaite sécurité au malade. La tendance actuelle est de soumettre les plans expérimentaux d’essais thérapeutiques à des comités d’éthique locaux, régionaux ou nationaux. Le Code d’éthique s’appuie sur les déclarations successives faites à Nuremberg (1947), à Helsinki (1964) et à T 拏ky 拏 (1977), sur les règles à suivre concernant les expérimentations cliniques, c’est-à-dire faites sur des sujets humains. La phrase introductrice de la déclaration de T 拏ky 拏 fixait la limitation de la poursuite de l’essai thérapeutique chez un sujet donné: «La mission du médecin est de veiller à la santé de l’homme.» Elle poursuivait: le serment de Genève engage le médecin à «considérer la santé du patient comme son premier souci», et le Code international d’éthique médicale «interdit au médecin de donner un conseil ou de poser un acte médical prophylactique, diagnostique ou thérapeutique qui ne soit pas justifié par l’intérêt direct du patient et notamment d’affaiblir la résistance physique ou mentale d’un être humain, à moins de nécessité thérapeutique». Dans cet esprit, la loi Huriet sur la recherche biomédicale fut adoptée par le Parlement français en décembre 1988.Essai ouvertLe principe en est simple. Le malade et le médecin connaissent le traitement utilisé. L’expérimentateur mesure les effets obtenus, soit par l’observation clinique, soit par des mesures biologiques. Il importe d’utiliser un grand nombre de sujets, car l’élément subjectif de la thérapeutique demeure entier tant du côté du malade que du côté du médecin. Le traitement statistique essaiera de mettre en évidence l’efficacité du traitement. Cette technique est le plus souvent utilisée lors des phases IV des essais. Ce type d’essais présente deux avantages dont le premier est la simplicité, la souplesse. Le second avantage réside dans la possibilité de modifier la thérapeutique à tout instant selon les réactions du malade. Cette méthode est parfaitement applicable quand on possède des moyens objectifs de mesurer l’effet du médicament. Elle est pratiquement la seule applicable pour des essais de très longue durée.Il existe également des inconvénients qui limitent l’extension de ce type de technique. Le premier est l’impossibilité de mesurer objectivement l’effet du médicament dans maintes maladies. En second lieu, il importe de ne pas méconnaître l’aspect psychologique de la prescription: connaissant le médicament prescrit, le médecin peut influencer la réaction du malade par son seul comportement. Le malade lui-même peut réagir de façon orientée, simplement parce qu’il sait recevoir un médicament nouveau.Essais contrôlésLes difficultés posées par la mesure objective des effets des médicaments, l’importance de la composante psychologique, la nécessité de comparer les effets obtenus avec un nouveau médicament, soit à un placebo, soit à un médicament connu, ont fait se développer les techniques dites d’essais contrôlés. Nous décrirons les trois méthodes principales des essais contrôlés.Méthode du simple aveugleLe médecin connaît, le malade ignore quel type de produit est administré. On supprime dans ce cas la composante psychologique du malade. Selon la maladie traitée et les possibilités techniques, on utilise simultanément le médicament nouveau et un médicament de référence ou un placebo. Pour tous les cas, les voies et le rythme d’administration sont parfaitement superposables. Les malades sont répartis au hasard en deux groupes, un groupe recevant le placebo ou le produit de référence, l’autre groupe recevant le produit nouveau. Dans cette méthode, si la composante psychologique du malade est diminuée, celle du médecin persiste. Applicable en phases II et III, seule à pouvoir être utilisée dans certains cas, c’est lorsqu’elle est associée à une possibilité de mesure objective de l’effet du médicament qu’elle prend toute sa valeur, car elle laisse au médecin toute liberté de modifier la prescription, comme il en serait si le médicament n’était pas en expérimentation. Elle permet en d’autres termes d’être proche d’une situation thérapeutique normale.Méthode du double aveugleLorsque l’impossibilité d’une mesure objective de l’effet thérapeutique ou le biais possible, lié au rôle psychologique du médecin, sont susceptibles de fausser les résultats, on emploie la méthode dite du double aveugle. Dans ce cas, le malade et le médecin prescripteur ignorent la qualité du produit administré. Comme dans la méthode précédente, la population tirée au sort a été séparée en deux groupes, l’un recevant le médicament témoin ou le placebo, l’autre, le produit nouveau. L’avantage majeur de cette technique réside dans la suppression complète des facteurs subjectifs d’appréciation de l’effet thérapeutique.L’inconvénient majeur est l’aspect artificiel de la situation thérapeutique. En effet, la sélection, la distribution au hasard de la thérapeutique, la non-connaissance par le médecin prescripteur de ce qu’il a prescrit est une situation qui n’existe pas en dehors de l’essai. Il devient difficile pour le médecin prescripteur d’adapter la thérapeutique à l’évolution de la maladie. Malgré tous ces inconvénients, la méthode du double aveugle est actuellement la méthode qui semble donner les meilleurs résultats pour l’essai de nouveaux médicaments, surtout quand il n’existe pas de méthodes objectives permettant de mesurer, de quantifier, l’effet thérapeutique.Méthode de l’analyse séquentielleCette méthode est une variante de la méthode du double aveugle. Elle permet, sur un plus petit nombre de malades et dans un délai de temps court, d’obtenir une réponse suffisamment précise, au début d’une phase III, pour confirmer l’intérêt que peut présenter le nouveau médicament. Les malades se servant de témoins à eux-mêmes reçoivent successivement le produit à l’essai et le produit de référence ou le placebo. On peut également apparier les malades, l’un servant de témoin à l’autre. On a préalablement pris le soin de définir statistiquement, sur un graphique, l’évolution probable en présence d’un traitement efficace, et on a défini les frontières de l’efficacité et de l’inefficacité. On porte ensuite sur ce graphique l’évolution des grandeurs biologiques ou cliniques mesurées, qui traduisent l’effet du traitement. Quand, au cours de l’évolution, la ligne coupe l’une ou l’autre des lignes préalablement tracées, on a la réponse à la question posée. L’inconvénient majeur de cette technique est qu’elle n’est applicable que dans les cas où l’on possède un élément simple et rapidement mesurable pour juger de l’effet de la thérapeutique.Autres méthodesNous avons à plusieurs reprises évoqué l’utilité d’une mesure objective de l’effet du médicament. Chaque fois que cette possibilité existe, associée et corrélée à la mesure des concentrations plasmatiques du nouveau médicament, elle remplit toutes les conditions d’un essai contrôlé. De plus, elle permet une surveillance thérapeutique réelle, identique à celle qui peut être exercée en dehors de l’essai clinique du nouveau médicament.Bilan de l’expérimentationIl ressort, à l’évidence, de ce qui précède que l’essai thérapeutique nécessite des techniques expérimentales dont la rigueur doit être au moins égale, sinon supérieure, aux techniques utilisées sur l’animal. Il est faux de croire que les impératifs éthiques de l’essai du médicament limitent la rigueur de l’expérimentation. Cette rigueur est l’une des composantes essentielles des précautions à prendre pour l’essai du médicament chez l’homme. Elle seule permet de traiter un malade, alors même qu’il reçoit un nouveau médicament. Le développement des méthodes modernes d’exploration diagnostique chez l’homme permet de développer cette rigueur, cette objectivité dans l’appréciation des effets.Le développement des méthodes statistiques spécifiquement appliquées à l’essai thérapeutique a permis de traiter correctement les résultats obtenus. Il reste le problème de l’appréciation du rapport risque/bénéfice. Il est des cas, favorables, où, à la suite des phases II et III de l’expérimentation, on a pu évaluer le rapport risque/bénéfice. Il est d’autres cas moins favorables où seule la phase IV permettra, à partir d’un grand nombre de malades traités pendant une longue période, d’évaluer ce rapport. Enfin, il ne faut pas oublier que, dans de nombreux cas, la connaissance physio-pathologique fragmentaire de la maladie traitée ne permet pas d’apprécier objectivement l’effet d’une nouvelle thérapeutique. Il reste dans ce domaine de nombreux progrès à réaliser qui permettront une amélioration des essais thérapeutiques.L’essai thérapeutique contribue par lui-même à améliorer la connaissance fondamentale du mécanisme des maladies et de l’effet des thérapeutiques, cela grâce à la rigueur introduite dans l’expérimentation. Cette étude doit être considérée comme une recherche fondamentale et doit bénéficier des mêmes efforts que d’autres recherches fondamentales.Enfin, c’est probablement lors de ces essais thérapeutiques que la relation entre le médecin et le malade peut s’exprimer avec le plus de force. La confiance du malade pour son médecin doit, en effet, être totale, et l’attention du médecin pour son malade doit être, s’il se peut, encore plus complète au cours de cet acte thérapeutique privilégié.5. PharmacovigilanceDéfinitionsLe médicament, c’est «toute substance ou produit utilisé ou destiné à être utilisé en vue de modifier ou d’étudier un système physiologique ou un état pathologique, dans l’intérêt du sujet auquel il est administré».La réaction adverse, c’est toute «réaction nocive, non recherchée, qui apparaît aux doses normalement utilisées chez l’homme», lors de l’utilisation thérapeutique d’un médicament.La pharmacovigilance (Adverse Drug Monitoring) a pour tâche d’«obtenir des indications systématiques sur les liens de causalité probables entre médicaments et réactions adverses dans une population». Son but est d’établir la liste des effets adverses des médicaments, c’est-à-dire de connaître de la façon le plus précise possible les effets dits secondaires, latéraux, imprévus ou indésirables (terme actuellement préféré en France), tous effets qui se développent parallèlement à l’effet thérapeutique recherché. Le travail de pharmacovigilance se situe après l’apparition de l’effet indésirable. Celui-ci est d’abord notifié par le clinicien qui l’a observé, il est ensuite étudié par l’équipe de pharmacovigilance, qui l’enregistre, en fait une première évaluation et le transmet aux instances nationales. Ce travail, réalisé à l’échelon national puis international, aboutit à une somme d’informations permettant de connaître tous les effets adverses d’un médicament et leurs fréquences relatives. Cette méthode s’apparente beaucoup aux techniques épidémiologiques, ce qui explique que l’on ait pu définir la pharmacovigilance comme l’«épidémiologie des effets adverses des médicaments». On comprend alors l’importance fondamentale de l’aspect systématique du recueil des données. C’est seulement si tous les cas d’effets adverses sont enregistrés que leur incidence sera connue avec précision. Les définitions du médicament et de la réaction adverse permettent de préciser le champ d’action de la pharmacovigilance.Les médicaments suivis sont déjà commercialisés; délivrés avec ou sans ordonnance; utilisés à des doses convenables; prescrits dans une indication convenable.C’est ainsi que les médicaments en cours d’expérimentation (expertises cliniques) ne relèvent pas actuellement de la pharmacovigilance. De même, les effets de doses toxiques ou résultant d’indications non thérapeutiques en sont écartés: ils relèvent de la toxicologie.Par rapport à ces textes, nous avons pris l’initiative de recueillir, outre les effets adverses, les effets imprévus mais bénéfiques (type action du clofibrate dans le diabète insipide). Bien que ceux-ci soient rares, ils sont en effet importants à connaître rapidement et peuvent représenter un atout supplémentaire pour le médicament dans une prescription ultérieure.Quelques particularités d’effets indésirablesIl peut paraître choquant d’admettre que les substances utilisées en thérapeutique, considérées comme des médicaments, reconnues officiellement comme tels, aient encore besoin d’être étudiées. Ne peut-on prévoir de façon complète et définitive les effets des médicaments avant leur commercialisation? Différents arguments permettent d’affirmer qu’un tel objectif relève de l’utopie. En effet, si certaines réactions adverses sont relativement simples à déceler, d’autres sont beaucoup plus difficiles à mettre en évidence.Ce sont d’abord les réactions adverses rares. Leurs fréquences sont de l’ordre de 10-6 ou 10-9. Or une étude préalable, même extrêmement approfondie, ne peut porter sur 106 ou 109 sujets; en outre, l’apparition d’un seul cas d’effet nocif dans la totalité de la population étudiée est difficilement interprétable en tant qu’effet adverse. Un tel effet est donc pratiquement imprévisible. Un exemple nous permettra de préciser cet aspect de certaines réactions adverses. La digitaline est utilisée en thérapeutique depuis 1785, d’abord sous forme de préparations de digitale, puis sous forme cristallisée (1868): c’est donc un très vieux médicament qui, entre autres effets adverses, peut provoquer des thrombocytopénies. À l’heure actuelle, on en connaît environ neuf cas, ce qui représente une fréquence de survenue extrêmement faible, compte tenu de l’utilisation de ce médicament. En supposant que la digitaline soit aujourd’hui en cours d’expérimentation, devrait-on attendre et prolonger celle-ci jusqu’à la caractérisation indiscutable des thrombocytopénies? Évidemment non, et devant l’intérêt que présentait cette molécule lors de ses premières utilisations, le risque plaquettaire, ignoré à l’époque, nous semble bien minime. Nous retrouvons ici la notion générale de risque thérapeutique inhérente à l’utilisation de tout médicament, qu’il faut prendre sans hésitation lorsque le bénéfice thérapeutique est évident, mais qui impose une surveillance constante (fig. 4).Les réactions adverses peuvent, dans certains cas, avoir une latence de survenue extrêmement longue, bien supérieure au temps des essais. Dans ces conditions, il est bien évident que seule la surveillance constante du médicament au cours de sa commercialisation permet de les dépister. Les exemples de ce type sont surtout ceux d’effets toxiques. Le cas des sels de thorium (radioactifs) utilisés comme opacifiants biliaires en 1930 est classique; ils peuvent entraîner, environ dix à quinze ans après une seule administration, l’apparition d’une atteinte néoplasique du foie et des voies biliaires ou d’une leucémie. Il est bien évident que si l’utilisation en clinique humaine de telles substances était plausible en 1930, elle ne le serait plus du tout aujourd’hui.En effet, d’une part, on n’utilise plus de substances radioactives de ce type, car leurs périodes sont trop longues (1,4 憐 1010 années pour le thorium), d’autre part, une étude métabolique (obligatoire actuellement) aurait montré que ces substances se fixent de façon pratiquement irréversible dans l’organisme, ce qui exclut tout intérêt thérapeutique. Cet exemple prouve cependant qu’il est possible d’observer aujourd’hui les effets d’une substance qui n’est pourtant plus utilisée depuis longtemps.Les réactions adverses peuvent aussi apparaître soudainement au cours de l’utilisation thérapeutique du médicament. Ces cas se différencient des précédents car ils ne peuvent être attribués à des latences de longue durée. L’exemple le plus actuel est celui des sels de bismuth qui, utilisés depuis le début du XIXe siècle, provoquent depuis environ cinq ans un nombre non négligeable d’encéphalopathies. Ces nouveaux effets adverses ont été signalés en Australie en 1973, puis en France en 1974. Un tel phénomème montre clairement la nécessité d’une surveillance constante de la population qui reçoit des médicaments.Comment expliquer l’extrême rareté de certains effets adverses comme les thrombocytopénies des digitaliques? Quelles sont les causes de l’apparition brutale et récente des encéphalopathies aux sels de bismuth?Dans le premier cas, il s’agit vraisemblablement de réactions immunologiques; dans le second, une «adaptation» récente de certaines bactéries intestinales aux sels de bismuth permettant la synthèse in situ de métabolites résorbables et toxiques a été invoquée. Dans les deux cas, pratiquement, on constate que ce sont moins les médicaments que leurs récepteurs – sujets sensibilisés à la digitaline, bactéries nouvelles capables de transformer les sels de bismuth en dérivés toxiques – qui sont en cause. Peut-être n’est-il pas inutile à ce propos de rappeler que la pharmacologie est une science biologique et que bon nombre de réactions imprévues aux médicaments ne s’expliquent que par la prodigieuse diversité de récepteurs qui caractérise l’espèce humaine. Ces quelques exemples montrent l’intérêt de la pharmacovigilance:– il est indispensable de recueillir les effets adverses des médicaments, c’est-à-dire de surveiller de façon permanente la population traitée;– les données obtenues n’ont qu’une valeur instantanée, elles doivent donc être systématiquement complétées;– du point de vue de la pharmacovigilance, l’étude d’un médicament n’est jamais terminée.Objectifs de la pharmacovigilanceCe sont d’abord la surveillance du prévisible et la recherche de l’imprévisible en matière d’effets médicamenteux. En d’autres termes, il s’agit à ce stade de surveiller de façon régulière l’importance des effets adverses déjà connus et, d’autre part, de déceler ceux qui n’avaient jamais été observés auparavant. À ce double objectif s’attache souvent une notion d’urgence: les accidents graves doivent être connus puis corrélés à la prise médicamenteuse le plus vite possible, ce qui suppose à la fois un véritable quadrillage de la population traitée et des moyens d’information rapides. De façon moins urgente et à plus long terme, l’objectif de la pharmacovigilance est de connaître, dès qu’elles deviennent significatives, toute variation de la fréquence d’un effet indésirable et toute apparition d’un nouvel effet adverse. La quantification des données utilise les méthodes épidémiologiques; c’est ainsi que l’on définit:– l’incidence d’un effet adverse, nombre de nouveaux cas observés pendant une certaine période;– la prévalence d’un effet adverse, nombre total de cas présents à un certain moment ou pendant une certaine période: c’est la somme des cas anciens persistants et des nouveaux cas.Ces valeurs, lorsqu’elles sont rapportées à l’ensemble de la population traitée, permettent de distinguer respectivement la fréquence des nouveaux cas et la fréquence instantanée d’un effet adverse. À titre d’exemple, le risque de survenue de quelques cytopénies médicamenteuses, évalué en Suède sur une période de cinq ans (1966-1970), se décompose comme le montre le tableau 1. Plus récemment, une augmentation importante des cas d’agranulocytose liés à l’administration d’un neuroleptique, la clozapine, a été constatée en Finlande (tabl. 2).Il est clair que de tous les pays où ce médicament est commercialisé, la Finlande a été la seule à présenter cette brutale augmentation de fréquence qui semble d’ailleurs s’être déclarée très rapidement, en quelques mois. Un facteur local de majoration de cet effet toxique bien connu est pratiquement certain; il a entraîné le retrait rapide (juill. 1975) de la clozapine du marché finlandais. Une telle décision ne se justifiait pas dans les autres pays où ce médicament est toujours utilisé sans que l’on ait observé jusqu’à présent d’augmentation de la fréquence des cas d’agranulocytose. Le mécanisme de survenue de cet effet n’est pas encore connu à l’heure présente.Si le premier objectif de la pharmacovigilance est de recueillir un maximum de données, le second est évidemment de pouvoir les redistribuer. Le retour au praticien est une étape fondamentale qui selon la gravité des cas peut revêtir deux formes principales:– l’alerte, dans toutes les éventualités d’effets toxiques majeurs, peut être donnée rapidement à chaque médecin et pharmacien, par l’intermédiaire de leurs ordres respectifs;– l’information régulière, qui doit être considérée comme une partie indispensable de l’enseignement postuniversitaire, n’est pas vraiment systématique, mais de nombreuses initiatives existent dans ce domaine.Enfin, les résultats obtenus servent de banque de données tant pour les pouvoirs publics que pour l’industrie pharmaceutique. Il y a là un système de référence dont la comparaison aux médicaments en expérimentation permettra de mieux préciser l’importance de leurs effets adverses et ainsi d’évaluer de façon plus rigoureuse un des aspects de leur intérêt thérapeutique.Les structures internationalesLe Centre international de pharmacovigilance de l’O.M.S. ou WHO drug monitoring center. Décidé à titre expérimental en 1963, le Centre mondial est définitivement créé en 1971 et Genève choisie comme lieu d’implantation. Les activités de ce centre sont multiples: centraliser les rapports que lui adressent les centres nationaux (Allemagne fédérale, Australie, Canada, États-Unis, France, Grande-Bretagne, Irlande, Nouvelle-Zélande, Israël, Japon, Pologne, Pays-Bas, Suède, Danemark, Norvège); les informations sont traitées à l’aide de systèmes informatisés, et une analyse mensuelle est adressée aux participants: harmoniser la terminologie, standardiser les critères d’évaluation des réactions indésirables, recevoir communication des décisions officielles prises par chaque pays membre concernant les médicaments dans le cadre de la pharmacovigilance et les transmettre aux autres pays; susciter des enquêtes au niveau des centres nationaux, qu’elles soient monocentriques ou multicentriques; provoquer des réunions d’experts et la publication de leurs travaux, en particulier en ce qui concerne la méthodologie. Les informations répercutées par ce centre international ne sont transmises qu’aux gouvernements des États participants, et ce confidentiellement, chaque gouvernement étant libre de les utiliser comme bon lui semble.Les centres nationaux de pharmacovigilance. Créés à l’instigation de l’O.M.S., ils se rapprochent plus ou moins de la structure type qu’elle préconise: une équipe permanente technique d’exécution, souvent composée de fonctionnaires de l’État aidés par des spécialistes scientifiques et médicaux, pharmaciens, épidémiologistes, statisticiens, informaticiens..., un comité national composé d’experts en médecine clinique, en pharmacologie, en toxicologie, en épidémiologie et en statistiques. Le rôle de chaque centre national devrait être triple: recueillir et évaluer les effets indésirables, organiser des enquêtes, enfin assurer l’information et le conseil thérapeutique.Les structures françaisesL’organisation de la pharmacovigilance en France remonte à l’arrêté promulgué le 2 décembre 1976 (Journal officiel du 19 décembre 1976) par le ministère de la Santé.Il avait pour objet la mise en place de trois structures de contrôle agissant en coordination. Des centres de pharmacovigilance hospitalière étaient institués dans les centres hospitaliers régionaux. Un Centre national de pharmacovigilance (C.N.P.V.) devait collationner les informations provenant de tous les secteurs de l’activité sanitaire. Auprès du ministre de la Santé était constituée une commission technique qui avait pour mission de proposer au ministre les décisions à prendre à la suite des constatations faites par les instances ayant recueilli les données.À ces structures, le décret du 24 mai 1986 a ajouté une quatrième instance, renforçant ainsi le dispositif comme indiqué ci-dessous.Le Centre national de pharmacovigilance a été créé en 1973, fondé par les membres du Conseil de l’ordre des médecins, le Conseil de l’ordre des pharmaciens, les centres anti-poisons, le Syndicat national de l’industrie pharmaceutique; il travaillait avec les centres hospitaliers de pharmacovigilance, son rôle a un peu changé depuis 1984.Les centres régionaux de pharmacovigilance tous placés, à la demande du ministère de la Santé, dans des centres hospitaliers et universitaires (C.H.U.) auprès d’un service de pharmacologie ou de centres de traitement des intoxications. Les centres régionaux de pharmacovigilance sont actuellement au nombre de 30. Le décret de 1984 demande aux médecins de déclarer obligatoirement les effets inattendus ou toxiques (effets indésirables, mauvais usages...) des médicaments qu’ils ont prescrits au centre de pharmacovigilance le plus proche. Les centres conduisent les enquêtes demandées par le ministre et enregistrent les données dans un système informatique commun. Ils ont également pour mission de diffuser l’information sur les médicaments.Le Comité technique de pharmacovigilance coordonne et évalue les enquêtes effectuées par les centres régionaux.La Commission nationale de pharmacovigilance apprécie les résultats de ces enquêtes et donne des avis au ministère de la Santé dont elle dépend directement sur les mesures à prendre pour protéger la santé publique (information, modification des conditions d’utilisation, retrait éventuel du marché).Parallèlement, les industriels titulaires d’une autorisation de mise sur le marché (A.M.M.) d’une spécialité pharmaceutique sont tenus de déclarer tous les effets inattendus ou toxiques de leur spécialité directement à la Direction de la pharmacie et du médicament qui assure le secrétariat du Comité technique et de la Commission nationale de pharmacovigilance. Les mesures décidées par le ministre chargé de la santé sont divulguées par voie de presse si elles sont importantes. Cette déclaration doit se faire tous les semestres la première année de la commercialisation ou annuellement ensuite. Les modifications se répercutent dans l’A.M.M. et la monographie de la spécialité concernée.La notification des effets adversesLa notification est assurée essentiellement par les médecins ou les pharmaciens, qui ont à leur disposition une fiche de recueil des données, analogue à celle qu’utilisent les médecins praticiens, et qui peuvent aussi faire parvenir ces notifications à la Commission nationale via l’ordre des pharmaciens. Les professions paramédicales citées dans l’article 3 de l’arrêté ont aussi la possibilité d’en utiliser.Dans le cadre hospitalier, le problème qui se pose actuellement est de savoir comment améliorer la notification des réactions adverses par tous les hôpitaux dans le cadre des centres hospitaliers de pharmacovigilance déjà existants, qu’il s’agisse de la surveillance systématique ou de la mise en garde immédiate.À qui incombe la notification d’un effet adverse observé au lit d’un malade? En premier lieu, elle revient au(x) médecin(s) responsable(s) du lit. Cette activité supplémentaire peut, à première vue, rebuter un médecin déjà très occupé. Plusieurs solutions sont possibles, dont la plus commode serait peut-être la désignation d’une personne du service (médecin, étudiant médecin, infirmière), responsable de prendre systématiquement contact avec le centre de pharmacovigilance local. Il nous semble également que les pharmaciens de chaque établissement hospitalier pourraient utilement seconder les cliniciens dans ce travail de notification. Il existe actuellement une fiche cartonnée, dite fiche d’alerte, très simple et très rapide à remplir, qui suffit à jouer son rôle de pense-bête auprès du médecin et de matériel de notification à adresser au centre local. Ce rapport très succinct sert de base pour l’établissement d’une fiche beaucoup plus détaillée, par un des membres du centre. Cette fiche, dite fiche en clair, est codée sur un document et envoyée au centre informatique du Comité technique de pharmacovigilance qui regroupe les données pour l’ensemble de la France.Tout effet secondaire, du plus fréquent au plus inattendu, doit être notifié. Il n’y a pas de critère de présélection dans les notifications à faire. Certes, il peut paraître inutile parce que très connu de signaler une atteinte gastrique avec de l’aspirine. Il se peut cependant, chez un sujet donné, qu’on s’aperçoive en étudiant l’anamnèse que l’aspirine était bien tolérée avant l’intervention de tel ou tel facteur concomitant. Ainsi, d’un faisceau d’observations peut se dégager un certain nombre de conditions qui pondèrent la responsabilité d’un médicament dans le déclenchement d’une pathologie iatrogène. Quand l’alerte est ainsi donnée, les cliniciens hospitaliers sont bien placés pour apporter une contribution précieuse à la pharmacovigilance en aidant à faire l’examen critique de la relation envisagée entre le médicament et la réaction adverse.Il est possible de procéder de deux manières pour recueillir les effets adverses. La première consiste, au cours des traitements, à noter tout effet adverse au fur et à mesure qu’il est constaté: c’est la pharmacovigilance extensive ou spontanée , dont la méthodologie s’apparente à l’épidémiologie descriptive. Dans ce genre de pharmacovigilance, tous les praticiens de l’aire géographique du centre sont concernés et la bonne marche du travail dépend d’abord d’eux. La qualité des notifications adressées au centre dépend en effet de leur bonne foi: les informations tronquées, dissimulées, ou omises involontairement faussent en effet le système dès le départ. Elle dépend aussi de leur compétence à identifier l’effet adverse, de l’intérêt qu’ils prennent à la question, du temps dont ils disposent. Discipline médicale nouvelle, presque encore au stade expérimental, du moins en France, elle ne fait pas encore l’objet d’un enseignement systématique dans les facultés de médecine. Ce système de recueil des données est limité par plusieurs facteurs. Les données recueillies par la notification spontanée restent numériquement trop faibles par rapport aux événements réels pour constituer un échantillon statistiquement valable; de plus, le nombre d’administrations du médicament (importance de sa consommation) n’est au mieux qu’approximativement connu. Il n’est donc pas possible de calculer une fréquence exacte de survenue, mais tout au plus de se faire une idée de l’importance du phénomène. De plus, la pharmacovigilance extensive n’apporte pas d’explication du mécanisme des accidents. Au total, la pharmacovigilance extensive permet de soupçonner la relation de cause à effet médicament-effet adverse, de regrouper des facteurs de corrélation éventuels: âge, sexe, ... de signaler des interactions médicamenteuses, et elle aboutit à formuler des hypothèses qui, si elles sont suffisamment argumentées et les faits envisagés suffisamment graves, peuvent justifier la prise de mesures d’urgence comme ce fut le cas en France avec les encéphalopathies mortelles après l’utilisation de sous-nitrates de bismuth.La seconde méthode de travail s’appelle la pharmacovigilance intensive , dont un des objectifs est de confirmer les données de la pharmacovigilance extensive. Elle étudie les problèmes de pharmacovigilance dans des limites définies de temps et/ou d’espace. Elle fait appel à des techniques de recueil systématique (et non plus spontané) des données. Elle peut se faire par malade, par médicament, par effet indésirable et, dans chacun de ces cadres, elle consiste à relever respectivement tous les effets adverses chez un groupe de malades, tous les effets adverses d’un certain type chez tous les malades quel que soit le médicament.Dans les méthodes dites numériques, on procède par des études prospectives directement sur une population, ou par des études rétrospectives, sur dossier, moins satisfaisantes, car elles comportent plus de biais importants. Dans les méthodes par estimations, on procède à l’étude des registres de mortalité et à l’estimation du risque ajouté par un médicament à celui qui existe déjà spontanément de voir survenir une pathologie donnée (par exemple, relation entre œstroprogestatifs et thrombophlébites).Dans les études numériques, on peut calculer avec précision une fréquence de survenue d’un effet et s’assurer de sa spécificité en utilisant une population témoin; dans les études par estimations, on isole le facteur par lequel la prise d’un médicament multiplie le risque de survenue de l’effet indésirable.Traitement de la notificationLe traitement de la notification comporte plusieurs étapes. D’abord, il faut préciser si la notification correspond bien à un effet indésirable, et, si oui, s’il est dû à la prise d’un ou de plusieurs médicaments (spécificité), établir sa gravité et sa fréquence, déterminer son mécanisme. Établir l’imputabilité d’un effet indésirable reconnu comme tel à l’action d’un médicament est une opération complexe, car elle requiert que soient réunis un certain nombre de critères pour qu’on puisse situer la responsabilité du médicament dans une appréciation globale dont les termes sont: certain, probable, possible, éventuel et enfin douteux. Chacun de ces termes correspond à la présence d’un certain nombre de critères d’imputabilité qui servent à l’étude analytique de la réaction indésirable et qui sont la chronologie de l’événement, la disparition des troubles lors de la réinduction du traitement, l’absence d’autres causes pouvant expliquer l’effet, l’existence d’examens complémentaires biologiques déterminants (dosage du médicament, par exemple), le caractère connu et non connu de l’effet indésirable (bibliographie). De plus, chacun des critères est évalué de 0 à 3: 0 = non recherché, 1 = négatif ou inconnu, 2 = intermédiaire ou douteux, 3 = positif ou évocateur. Le problème de l’imputabilité est compliqué par le fait des associations médicamenteuses soit à l’intérieur de la même forme galénique, soit par la polymédication. À partir du moment où l’effet adverse est identifié comme tel, et évalué dans son rapport avec le traitement médicamenteux, sa fréquence, sa gravité, il est transmis, en France, par le centre de pharmacovigilance référencé (c’est-à-dire local) qui en a fait l’étude à un centre de traitement informatisé des données de pharmacovigilance créé et situé à Lyon, qui permet une mise en mémoire de toutes les notifications recueillies en France. Cette transmission se fait à l’aide du dossier codé à partir de l’observation prise en langage clair. Le codage est réalisé par les équipes des centres à l’aide de dictionnaires qui permettent une standardisation des entrées des données et qui sont un dictionnaire des médicaments et un dictionnaire des symptômes et des réactions adverses. L’accès à ces données informatisées est régi par une convention tripartite: ministère de la Santé, Association des unités de pharmacovigilance hospitalière, structure informatique lyonnaise. Cette convention garantit l’accès limité à un personnel de pharmacovigilance et officiellement reconnu. Ce centre informatique français transmet ses données à celui de l’O.M.S., dont l’ordinateur garde en mémoire l’ensemble des informations et adresse tous les trois mois aux pays participants un rapport qui comporte: toutes les réactions notifiées, classées par noms de médicaments; tous les médicaments mentionnés, classés par effets adverses présumés; tous les effets adverses signalés pour la première fois; la liste des réactions létales et les malformations congénitales, des «signaux» attirant l’attention sur les augmentations au-dessus d’un certain seuil de fréquence au cours de la dernière période; des rapports concernant un effet indésirable ou un médicament relativement à leur fréquence dans la mémoire générale. Chaque rapport porte sur les derniers trois mois. À l’heure actuelle, les notifications faites par le secteur médical non hospitalier sont de plus en plus fréquemment adressées aux centres hospitaliers de pharmacovigilance qui les regoupent. Les réflexions suggérées par les problèmes de recueil et de traitement des données par les centres s’appliquent donc également à la pharmacovigilance non hospitalière.
Encyclopédie Universelle. 2012.